All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a Healthcare Professional. If you are a patient or carer, please visit the Lymphoma Coalition.
The lym Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the lym Hub cannot guarantee the accuracy of translated content. The lym and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


The Lymphoma & CLL Hub is an independent medical education platform, sponsored by AbbVie, BeOne Medicines, Miltenyi Biomedicine, Nurix Therapeutics, Roche, Sobi, and Thermo Fisher Scientific and supported through educational grants from Bristol Myers Squibb, Lilly, and Pfizer. Funders are allowed no direct influence on our content. The levels of sponsorship listed are reflective of the amount of funding given. View funders.
Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account and access these new features:
Bookmark content to read later
Select your specific areas of interest
View lymphoma & CLL content recommended for you
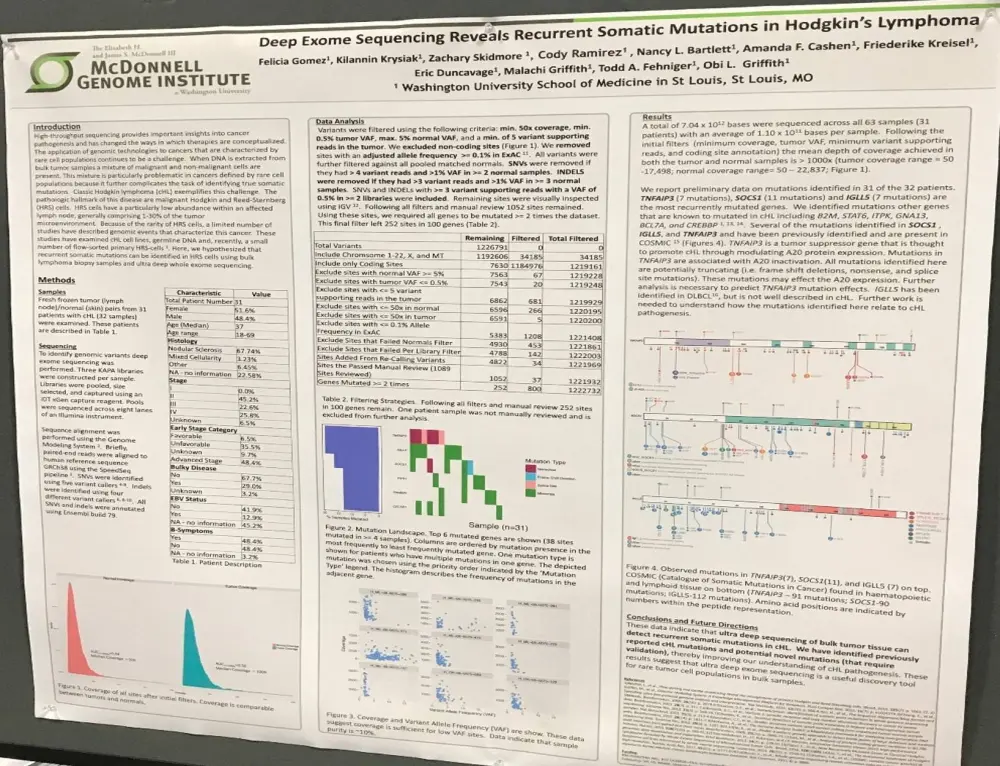
AACR 2017 | Poster 2448/5 – Deep exome sequencing reveals recurrent somatic mutations in Classical Hodgkin Lymphoma
On Monday 3rd April during this year’s American Association for Cancer Research (AACR) annual meeting, a poster (2448 / 5) by Felicia Gomez, from Washington University, St. Louis, MO, et al. titled “Deep exome sequencing reveals recurrent somatic mutations in Hodgkin's Lymphoma” was presented.
The group hypothesized that recurrent somatic mutations can be recognized in Hodgkin Reed-Sternberg (HRS) cells, taken from biopsies of bulk Hodgkin Lymphoma (HL), using ultra-deep exome sequencing.
Key Highlights:
- In total, using 63 samples from 31 patients, 7.04x1012 bases were sequenced; 1.10x1011 average bases per sample
- Mean depth of coverage achieved in both tumor and normal samples is >1,000x; tumor coverage range = 50–17,498; normal coverage range = 50–22,837
- Mutations identified in 31/32 patients
- The most currently mutated genes = SOCS1 (11 mutations), and TNFAIP3 and IGLL5 (7 mutations each)
- Mutations in TNFAIP3 were frameshift deletions, nonsense, and splice site mutations (all potentially truncating)
- Identified other mutations known to be mutated in cHL = B2M, STAT6, ITPK, GNA13, BCL7A, and CREBBP
The poster concluded by stating that recurrent mutations in cHL can be identified using ultra-deep sequencing of bulk tumor tissue. The group identified previously unreported mutations which require validation, as well as known mutations. This data improves our understanding of the pathogenesis of cHL.
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content