All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a Healthcare Professional. If you are a patient or carer, please visit the Lymphoma Coalition.
The lym Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the lym Hub cannot guarantee the accuracy of translated content. The lym and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


The Lymphoma & CLL Hub is an independent medical education platform, sponsored by AbbVie, BeOne Medicines, Miltenyi Biomedicine, Nurix Therapeutics, Roche, Sobi, and Thermo Fisher Scientific and supported through educational grants from Bristol Myers Squibb, Lilly, and Pfizer. Funders are allowed no direct influence on our content. The levels of sponsorship listed are reflective of the amount of funding given. View funders.
Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account and access these new features:
Bookmark content to read later
Select your specific areas of interest
View lymphoma & CLL content recommended for you
iwCLL 2017 | What is the cell of origin of Chronic Lymphocytic Leukemia?
On Monday 13th May, the first session during iwCLL 2017 was titled “Unraveling the factors leading to the development of CLL”, and was chaired by Daniel Catovsky (Institute of Cancer Research, London, UK) and Stephan Stilgenbauer (University of Ulm, Germany).
A talk titled “What is the cell of origin of CLL?” was presented during this session by Olivier A. Bernard, PhD, from the Institut Gustave Roussy, Paris, France.
So far, from immunophenotype and transcription profile analyses, Seifert et al. have reported that unmutated IGHV CLL derives from unmutated mature CD5+ B-cells and mutated IGHV CLL derives from a distinct CD5+CD27+ post-germinal center B-cell subset. Moreover, Kikushige et al. have found that hematopoietic stem/progenitor cells from patients with CLL can engraft into immunodeficient mice and display cell-intrinsic propensity to generate clonal CLL-like B-cells, restricted always to mono- or oligo-clones. However, Bernard asked if we can identify the cell in which the first acquired mutation occurs?
In 24 samples from a French protocol, flow sorting was carried out for CD3+, CD19+(CD5), CD14+, and CD34+ on cells from blood samples (>96% purity). In myeloid conditions, single CD34+,CD19- cells were grown in vitro. Acquired coding sequence mutations were identified by comparing tumor and CD3+ DNA. Mutation frequencies were evaluated in sorted populations and genotype was assessed in colonies from CD34+ single cells.
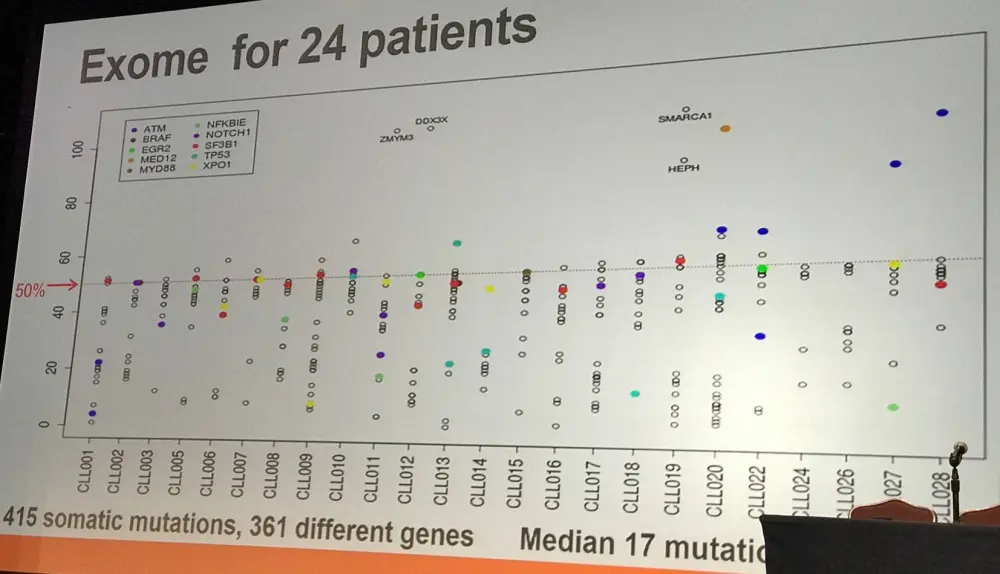
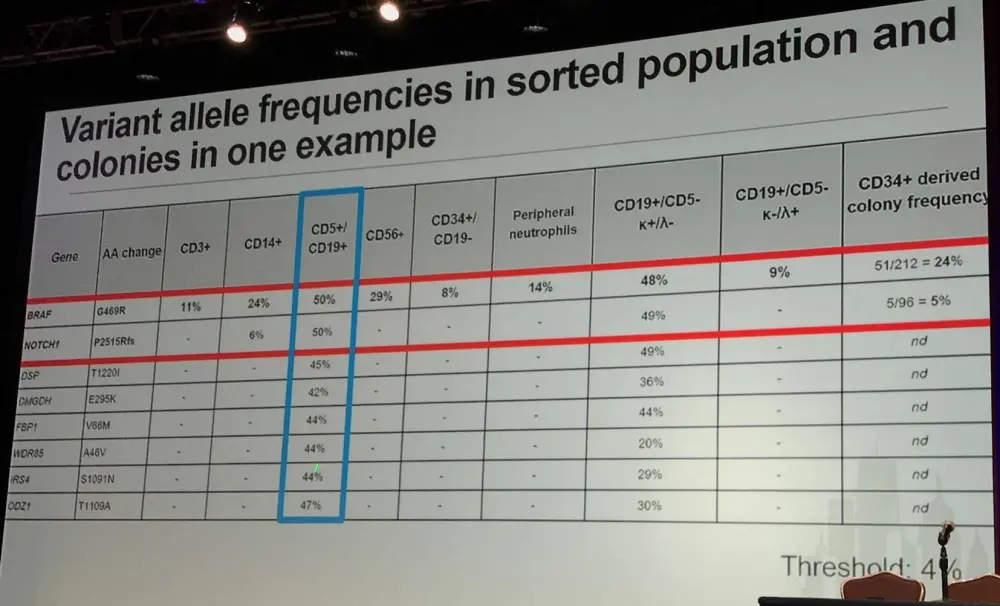
In the first group identified (14/24 samples; 58%), mutations were found in all CD34+, CD14+, and CD19+ fractions. They displayed normal differentiation (lymphoid and myeloid) of the mutated CD34+ progenitors.
In the second group identified (7/24 samples; 29%), mutations were found in CD34+ and CD19+ but not CD14+ fractions. Moreover, a bias was found toward lymphoid differentiation.
In the third group identified (3/24 samples; 13%), mutations were found only in the CD19+ fraction. Additionally, there was a strict commitment found towards lymphoid differentiation and cells displayed other mutations (non-coding, genomic deletion, gain, etc.)
In progenitors, most mutated genes are oncogenes and have been found to be recurrently mutated in hematological malignancies. These “early” mutations drive clonal dominance; however, Bernard asked if they affect B-cell differentiation/maturation?
|
(1: Damm et al. 2014; 2: Mansouri et al. 2015; 3: Young et al. 2017). |
|
|
NOTCH1 |
5–15% in CLL |
|---|---|
|
SF3B1 |
5–20% |
|
MED12 |
2–5% |
|
NFKBIE1,2 |
5–10% |
|
MLL2 |
1–2% |
|
XPO1 |
2–4% |
|
BRAF |
3–4% |
|
EGR21,3 |
2–8% |
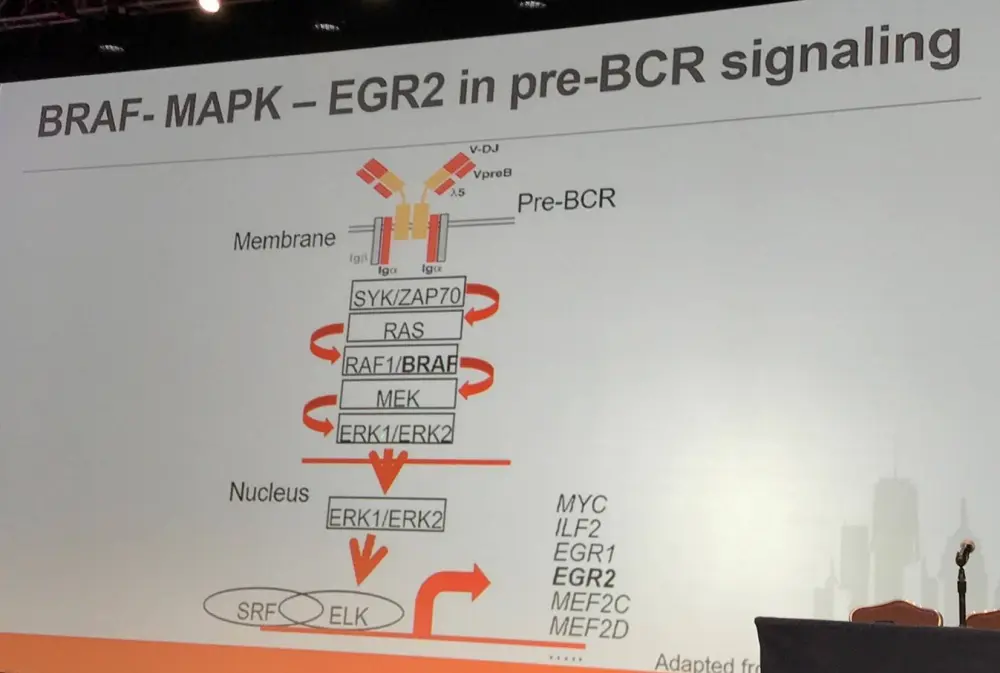
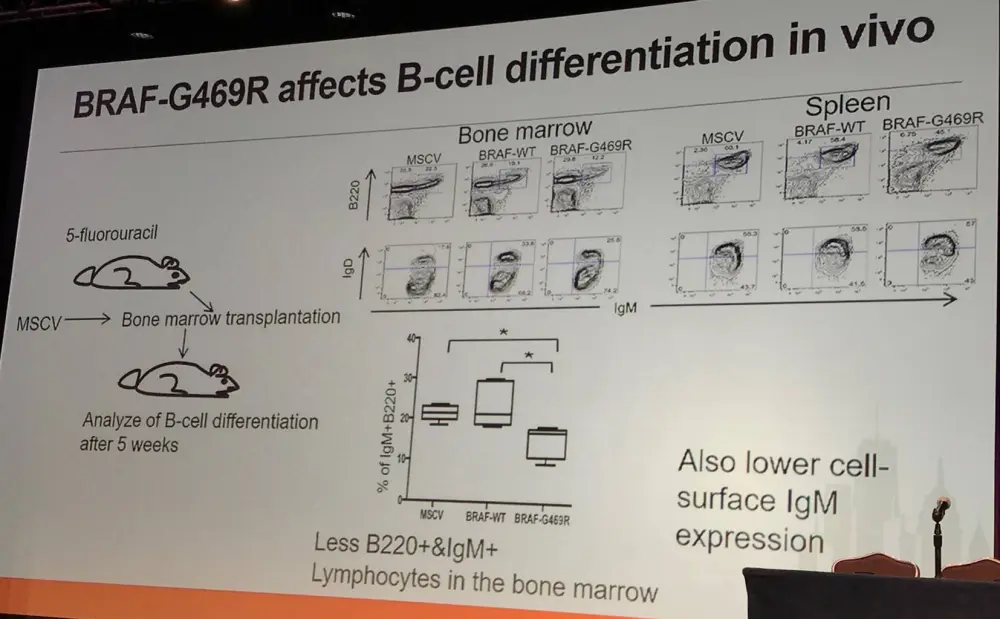
It has been reported by Damm et al. that BRAF-G469R constitutively phosphorylates ERK, activating Egr2 transcription in Ba/F3 cells (IL-3 dependent murine pro-B-cell line). Moreover, in a murine model, BRAF-G469R was found to affect differentiation:
Additionally, it has been found that mutant EGR2 and activated B-Cell Receptor (BCR) signatures overlap. When comparing EGR2-E356K to EGR2-WT, 224 gene signatures were found to be overexpressed (only 15 were downregulated), and 168 were direct EGR2-targets. Differential expression of EGR2 has resulted in enrichment of BCR-stimulated normal B-cells (Vallat et al. 2007). Also, genes upregulated upon BCR stimulation of normal B-cells are enriched in EGR2-mutant CLL.
Olivier Bernard concluded the talk by stating that CLL develops from a pre-leukemic phase, similar to many adult myeloid malignancies as well as Hairy-Cell Leukemia, B-Cell and Angioimmunoblastic T-Cell Lymphoma. Additionally, “early” mutations in CLL affect B-cell differentiation/maturation as well as contribute to growth/survival of the tumor clone.
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content