All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a Healthcare Professional. If you are a patient or carer, please visit the Lymphoma Coalition.
The Lymphoma Hub uses cookies on this website. They help us give you the best online experience. By continuing to use our website without changing your cookie settings, you agree to our use of cookies in accordance with our updated Cookie Policy
Introducing

Now you can personalise
your Lymphoma Hub experience!
Bookmark content to read later
Select your specific areas of interest
View content recommended for you
Find out moreThe Lymphoma Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the Lymphoma Hub cannot guarantee the accuracy of translated content. The Lymphoma Hub and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.









EHA 2017 | Molecular biology and treatment of Waldenström Macroglobulinemia
Bookmark this article
While at the 22nd Congress of the European Hematology Association (EHA) in Madrid, Spain, the LH attended a session on Waldenström Macroglobulinemia (WM), chaired by M.V. Mateos from the University Hospital of Salamanca, Spain.
Molecular Biology of Waldenström’s disease
The first of two talks during this session was given by Steven P. Treon, MD, PhD, from Harvard Medical School, Boston, US.
Until recently, treatment strategy for WM has been a “hand me down” approach – wait for strategies to be explored and defined in other Lymphoma subtypes and then use those. Whole genome sequencing led to MYD88 L265P detection (affects the TIR domain responsible for dimerization), which is present in around 95% of WM patients. The mutation results in permanent dimerization and causes constitutive activation of the Bruton’s Tyrosine Kinase (BTK) pathway via Toll-Like Receptors (TLRs). Only around 2% of cases harbor non-L265P MYD88 mutations. MYD88 mutations have also been found to transactivate NF-kB.
Ibrutinib, a pleiotropic BTK inhibitor, has been approved by the U.S Food and Drug Administration (FDA) as well as the European Medicines Agency (EMA) for WM. Hematopoietic Cell Kinase (HCK), a key component of ibrutinib targeting, is normally downregulated after differentiation, but is upregulated in WM cells. Mutated MYD88 over-expression results in HCK and IL-6 transcription, whereas HCK knockdown decreases survival and attenuates BTK, PI3K/AKT, and mitogen-activated protein kinase/extracellular signal-regulated kinase signaling in mutated MYD88 WM. Ibrutinib and A419259, a more potent HCK inhibitor, blocked HCK activation and induced apoptosis in mutated MYD88 WM cells. Docking and pull-down studies confirmed that HCK was a target of ibrutinib. Ibrutinib and A419259 also blocked adenosine triphosphate binding to HCK, whereas transduction of T333M (HCK gatekeeper mutant) in mutated MYD88 expressing WM cells promotes resistance to ibrutinib and A419259. Therefore, these findings suggest that expression and activation of HCK is caused by mutated MYD88, supports the growth and survival of mutated MYD88 WM cells, and is a direct target of ibrutinib. HCK presents as a potential novel therapeutic target for MYD88-mutated WM (Yang et al. Blood. 2016).
Next, Treon discussed CXC Chemokine Receptor 4 (CXCR4) C-tail mutations, which occur in around 30–40% of WM patients and are very rate in those with other lymphoproliferative disorders. More than 30 nonsense and frameshift mutations have been identified. CXCR4 c-tail mutations work downstream of MYD88 mutations and prevent correct localization of GRK2/3, promoting resistance to ibrutinib via enhancing AKT/ERK signaling. Ulocuplumab is a monoclonal antibody which targets CXCR4 and has potential antineoplastic activity. It prevents the binding of Stromal Derived Factor-1 (SDF-1) to the CXCR4 receptor and subsequent receptor activation, potentially causing a reduction in tumor cell proliferation and migration. A phase II trial of ibrutinib (420mg PO daily) plus ulocuplumab (x4 weekly then biweekly x20) in patients with CXCR4WHIM WM patients has been planned.
Furthermore, it has been found that in WM patients treated with ibrutinib, signaling via the IRAK1/4 kinase pathway remains intact in resistant cells. Both the BTK and IRAK1/4 pathways result in NF-kB signaling.
Treon then moved on to discuss BCL-2 in WM:
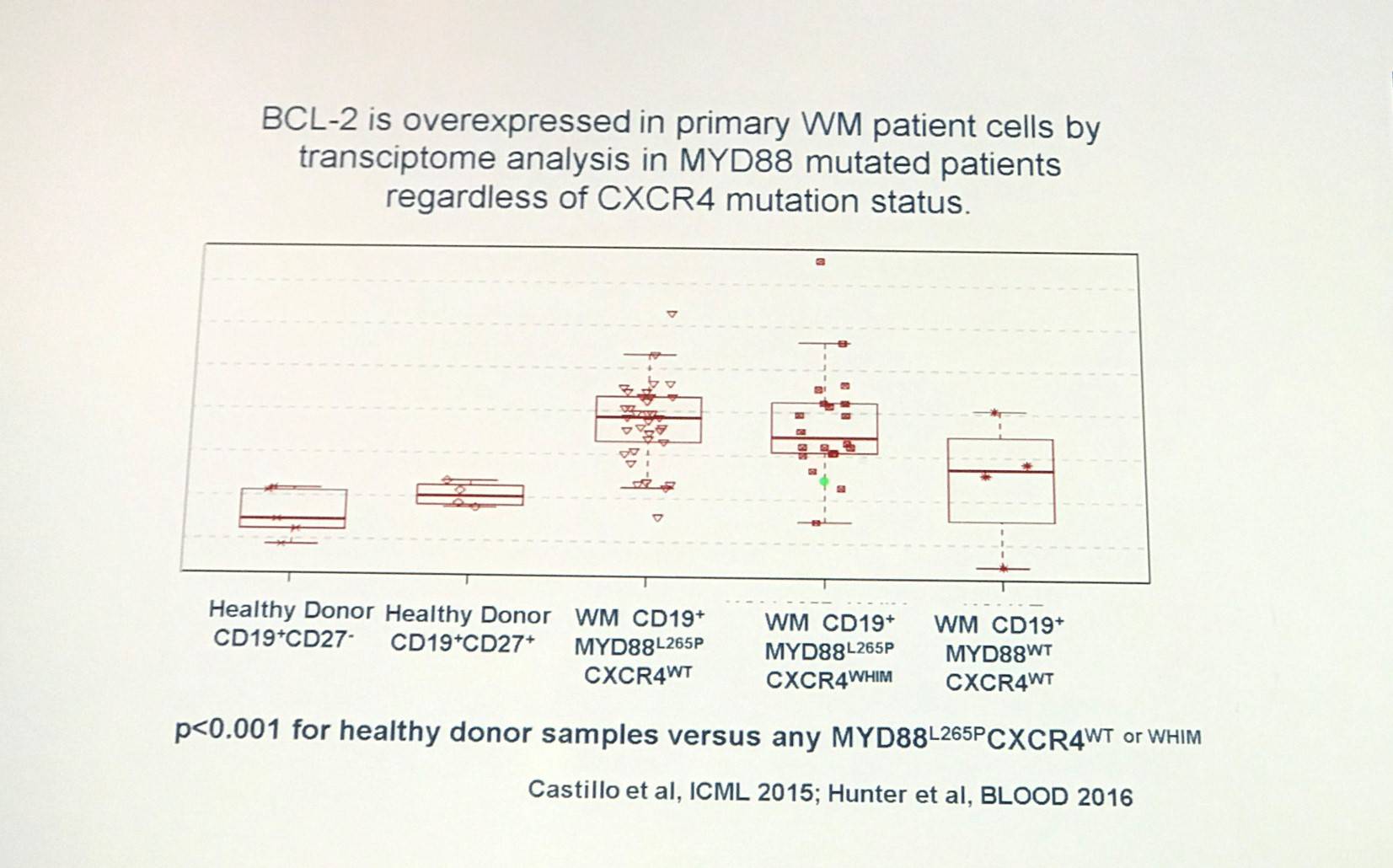
It has been found that venetoxlac (ABT-199), a selective small molecule inhibitor of BCL-2, enhances the activity of ibrutinib in MYD88 mutated WM cells (Cao et al. BJH. 2015).
Treon concluded his half of the session by stating that MYD88 and CXCR4 mutations are common in WM. MYD88 activates BTK and HCK in WM cells. Ibrutinib has been found to not only target BTK, but HCK too, and has achieved high response rates and durable responses in Relapsed/Refractory (R/R) WM. Furthermore, CXCR4 mutations promote resistance to ibrutinib that can be overcome by CXCR4 antagonists such as ulocuplumab. Multiple BTK mutations common with resistance to ibrutinib are associated with mutated CXCR4. Novel treatment options for WM include agents that target MYD88, CXCR4, and BCL-2 signaling.
Novel treatment strategies in Waldenström’s Macroglobulinemia
Efstathios Kastritis, MD, from the National and Kapodistrian University of Athens, Greece, presented the second talk in this session focused on novel treatment for WM.
Anti-CD20 monoclonal antibodies
The talk began by looking at anti-CD20 monoclonal antibodies for WM. We have had extensive experience with rituximab for WM of more than 15 years; some of the newer anti-CD20 antibodies include ofatumumab and obinutuzumab. Anti-CD20 antibodies are associate with low toxicity and no risk of Myelodysplastic Syndrome (MDS). They do have significant activity, but are slow. Thus far, no Complete Responses (CRs) have been reported with them for WM but they can be combined with nearly all other available agents. They can result in a transient increase of IgM, known as IgM flare, in around 40–60% of patients. Rituximab especially is associated with IgM neuropathy, cryoglobulins, and cold agglutinins and so prophylaxis should be considered. Kastritis gave a summary of first-line therapy for WM using rituximab-based strategies:
|
Regimen |
ORR |
VGPR/CR |
|---|---|---|
|
Rituximab x4 |
25–30% |
0–5% |
|
Rituximab x8 |
40–45% |
5–10% |
|
Rituximab/Thalidomide |
70% |
10% |
|
Rituximab/Cyclophosphamide i.e. R-CHOP, -CVP, CPR, DRC |
70–80% |
20–25% |
|
Rituximab/Nucleoside analogues i.e. FR, FC, CDA-R |
70–90% |
20–30% |
|
Rituximab/Bortezomib i.e. BD, VR |
70–90% |
30–40% |
|
Rituximab/Bendamustine (BR) |
90% |
30–40% |
In a phase II study of dexamethasone, rituximab, and cyclophosphamide (BRC) for WM (NCT01788020) with a minimum follow-up of 7 years, 83% in the Intent to Treat (ITT) analysis achieved a response: CR 7%, PR 67%, minor remission 9%. Stable Disease (SD) was reported in 9% and 8% experienced Progressive Disease (PD). Median Progression-Free Survival (PFS) was 35 months (95% CI, 22–48); disease progression at 3 years was 45%, and unrelated death, without disease progression, was 12%. Median time to next treatment was 51 months. Median Overall Survival was approximately 8 years (Kastritis et al. Blood. 2015).
A sub-analysis of WM patients in the StiL NHL1 study of BR compared to R-CHOP as first-line treatment was then presented (Rummel et al. Lancet. 2013). In 41 evaluable patients, response rate to BR and R-CHOP was 95% in both arms (21/22 and 8/19 patients, respectively). Median PFS in the BR arm was 69.5 months compared to 28.1 months in the R-CHOP arm (HR, 0.33; 95% CI, 0.11–0.64; P = 0.0033). Median OS at 6 years for BR and R-CHOP was 90.4% and 58.3%, respectively.
BR has also been compared to DRC in a retrospective analysis (Paludo et al. ASH. 2016. Abstract #2968). In first-line patients, the median time to best response for BR and DRC was 6.1 months (range, 1–25) and 11 months (range, 0.5–47), respectively (P = 0.13). Overall Response Rate (ORR) for BR patients was 93% (VGPR 29%, PR 57%, MR 7%) and 1 patient (7%) had PD. ORR for DRC patients was 96% (VGPR 17%, PR 70%, MR 9%) and 1 patient (4%) achieved SD. 2-year PFS for BR and DRC was 88% and 61%, respectively (P = 0.08). 2-year time-to-next therapy was 88% and 76% in the BR and DRC groups, respectively (P = 0.35). In R/R patients, median time to best response was similar for BR and DRC treated patients: 7 months (range, 1–39) and 7 months (range, 0.5–28), respectively (P = 0.77). ORR for BR patients was 95% (CR 3%, VGPR 38%, PR 41%, MR 13%). Two patients (5%) had PD. ORR with DRC was 87% (VGPR 4%, PR 64%, MR 19%). Four patients (9%) achieved SD and 2 patients (4%) had PD. 2-year PFS was 66% and 53% in the BR and DRC groups, respectively (P = 0.08). 2-year time to next treatment was 75% and 68% in the BR and DRC groups, respectively (P = 0.24).
A phase II, open-label, single-arm study (NCT00811733) of ofatumumab for WM was also discussed (Furman et al. Lancet Haematol. 2016). Overall, 37 patients were enrolled and assigned to treatment: 15 in treatment group A (300mg week 1 then 1,000mg weeks 2–4) and 22 in treatment group B (300mg week 1 then 2,000mg weeks 2–5). All 37 were included in the efficacy and safety analyses. An overall response after cycle 1 was achieved in 19 patients (51%; 95% CI, 34.4–68.1) and 22 patients (59%; 95% CI, 42.1–75.2) achieved an overall response after the re-dosing cycle; 15 (41%) with PRs, 7 (19%) with minor responses. The second treatment cycle was completed in 13 patients; 10 (77%) achieved a response. ORR was around 59% for all patients, 52% is rituximab exposed patients, and 75% in rituximab naïve patients. Median PFS was approximately 17.5 months and median time to response was around 2.5 months.
Proteasome inhibitors
Kastritis moved on to then discuss proteasome inhibitors for WM; they may have additive/synergistic effect with rituximab. In particular, bortezomib was looked at in detail:

The results from a phase II study of a chemotherapy free regimen of bortezomib, dexamethasone, and rituximab (BDR) with a median follow-up of 7 years in patients with newly diagnosed WM were discussed (NCT00981708). BDR was associated with high response rates and long-term remissions; median PFS was found to be 43 months and 7-year OS was 66%. So far, 20 patients (34%) have dies and 9 of these (15%) were from unrelated causes (Gavriatopoulou et al. Blood. 2017).
Carfilzomib, rituximab, and dexamethasone (CARD) was also covered in the form of a phase II trial (NCT01470196) of 31 WM patients (28 newly diagnosed; 3 rituximab, chemo, and PI naïve). MYD88 L265P was reported in 29 of 30 patients, and CXCR4WHIM was seen in 11 of 30 patients. ORR was 87.1%, with a CR of 3.2%, VGPR of 32.2%, PR of 32.2%, and MR of 19.3%. Median time to major response or better was 2.1 months and median time to best response was 12.81 months. CXCR4 status did not affect ORR. IgM flar was reported in 5 of 22 patients (Treon et al. Blood. 2014).
Carfilzomib and dexamethasone without rituximab was also looked at. In an experience of 7 patients (of which 6 had prior bortezomib, and 2 were bortezomib-refractory [both responded]), 3 patients achieved a PR, 2 achieved a VGPR, 1 achieved stringent CR, and there was 1 minor response. Median time to response was 2.4 months (Vesole et al. Leuk & Lymph. 2017).
Moreover, a phase II trial of ixazomib, dexamethasone, and rituximab in 26 patients with newly diagnosed WM was discussed (NCT02400437). An ORR of 88% was reported (VGPR 6%, PR 44%, MR 38%), with a major response rate of 50%. VGPR plus PR was 47% in CXCR4WHIM patients compared to 64% in patients with the wildtype gene (P = 0.32). Median time to response in all patients was 8 weeks; in CXCR4 mutated and unmutated patients was 12 weeks and 8 weeks, respectively (P = 0.03; Castillo et al. ASH. 2016. Abstract #2956).
A phase Ib/II trial has also been conducted of two different dosing schedules (2/7 or 5/14) of single-agent oprozomib in patients with WM (NCT01416428). In the phase Ib group, the 2/7 (n=8) and 5/14 (n=11) schedules achieved ORRs of 38% and 73%, respectively. In the phase II group (n=17), the 5/14 schedule (all patients) achieved an ORR of 59%. In carfilzomib naïve patients (n=16), ORR was 56%; in bortezomib refractory patients (n=3), ORR was 67%. The most common grade 3–4 Adverse Events were gastrointestinal (Siegel et al. ASH. 2014. Abstract #1715).
Anti-BTK-based therapy
The next class of agents discussed for WM were BTK inhibitors, which block BCR signaling mediated by mutated MYD88. Ibrutinib, the first-in-class BTK inhibitor, is orally administered, can be continually dosed, and has relatively low toxicity. There are new BTK inhibitors in clinical development for WM, such as acalabrutinib and BGB-3111.
In a phase II trial of ibrutinib in R/R WM, 3-year PFS, EFS, and OS were 82%, 68%, and 90%, respectively. Further results were stratified by MYD88 and CXCR4 mutation status (Treon et al. NEJM. 2015):
|
|
MYD88 L265P CXCR4 wildtype |
MYD88 L265P CXCR4 WHIM |
MYD88 wildtype CXCR4 wildtype |
P value |
|---|---|---|---|---|
|
N |
34 |
21 |
7 |
|
|
Overall RR |
100% |
80.9% |
57.1% |
<0.01 |
|
Major RR |
88.2% |
57.1% |
28.6% |
<0.01 |
Following this, the phase III iNNOVATE trial of ibrutinib in patients with rituximab-refractory WM was looked at in detail (NCT02165397).
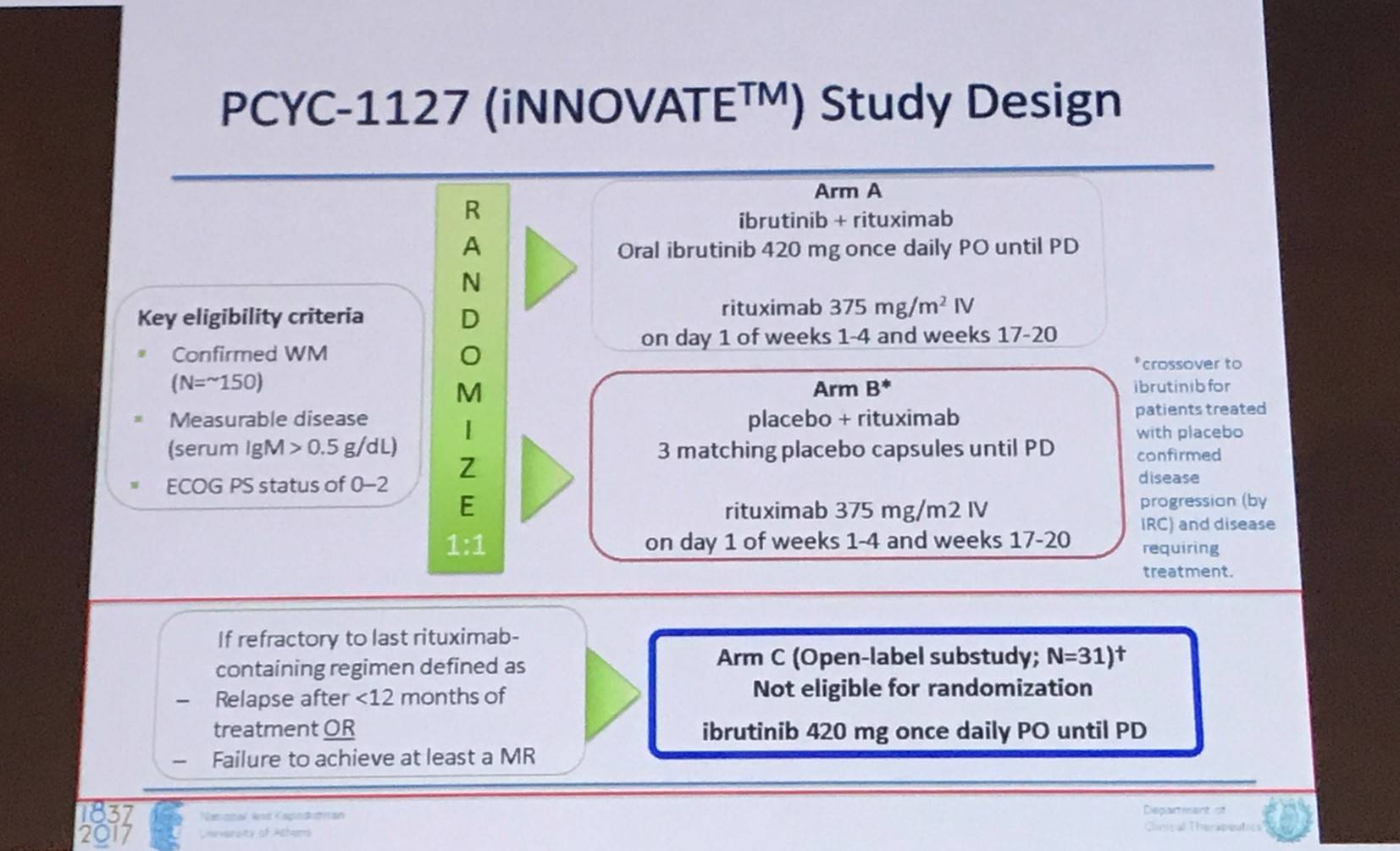
In all 31 patients, ORR was 90% with 4 patients achieving VGPR, 18 PR, and 6 MR. In MYD88 L265P / CXCR4 wildtype patients, ORR was 88% with 3 patients achieving VGPR, 11 PR, and 1 MR. In MYD88 L265P / CXCR4WHIM, ORR was 100% with 5 PRs and 2 MRs. 18-month PFS and OS was 86% and 97%, respectively (Dimopoulos et al. Lancet Oncol. 2016).
The new BTK inhibitor BGB-3111 was briefly mentioned; in a phase I/II trial in R/R WM, ≥PR was achieved in 83% (20/24) of patients, with a VGPR of 33% (8/24). The median time to initial response and major response were 29 and 34 days, respectively (Tam et al. ASH. 2016. Abstract #1216). A phase III trial comparing BGB-3111 and ibrutinib in patients with WM is currently ongoing (NCT03053440). During ICML 2017, updated safety and efficacy results from a cohort of WM patients treated with BGB-3111 in an ongoing open-label, multicenter, dose-finding, phase I study were presented (abstract #059); read more here.
Other new treatments
Kastritis began this portion of the talk by focusing on the mTOR inhibitor everolimus combined with bortezomib, and rituximab (RVR). In a phase I/II trial, 36/46 full doses of all three drugs were administered. For FDT ORR was 89% and PFS was 21 months. No dose limiting toxicities were reported, or deaths. The most common toxicities were fatigue (63%), anemia and leukopenia (52% each), neutropenia (48%), diarrhea (43%), as well as neuropathy and pneumonitis/pulmonary infiltrates (41% each; Ghobrial et al. Leukemia. 2015).
Furthermore, a phase I study of venetoclax in patients with R/R NHL, 4 patients had WM (median 4 prior lines). All 4 patients achieved a PR. Median time to first response was 74.5 days, and DoR for the 4 WM patients was 11.1, 12.4, 38.2, and 41.5 months (Davids et al. JCO. 2017). A phase II study of R/R WM is ongoing (NCT02677324).
Conclusion
Kastritis rounded off the session by focusing on when it is necessary to being treatment, as not all patients with a diagnosis of WM require immediate therapy. The criteria for the initiation of therapy are:
- IgM-related complications (e.g. hyperviscosity, amyloidosis, hemolysis, symptomatic cryoglobulinemia, moderate-sever IgM-related neuropathy)
- Symptoms related to direct BM involvement by tumor cells or tumor bulk (e.g. cytopenias, constitutional symptoms, bulky extramedullary disease)
It was emphasized that IgM levels alone are not an indication for initiation of therapy. WM is a disease of the elderly and so management of symptoms can be sufficient to prolong life, however toxicity is a primary concern and “one size does not fit all”. Managing symptoms is not sufficient in younger patients and those with complications associated with low levels of paraprotein. Treatment of WM requires consideration of disease (bulk, IgM levels, presence of cytopenia, immunologic complications) and patient (age, comorbidities, oral vs. parenteral therapy, candidacy of ASCT) characteristics. Additionally, there are also treatment-related factors to consider in terms of activity (short term vs. long term), toxicity (hematologic and non-hematologic), and late complications (MDS, secondary primary malignancies, and transformation to DLBCL). Some new therapeutic strategies were also mentioned, including anti-CD38 monoclonal antibodies, checkpoint inhibitors (e.g. PD-1 is expressed in WM cells), and Chimeric Antigen Receptor (CAR) T-cells targeting CD19 (expressed in LPLs).
For now, in 2017, anti-CD20 therapy remains a primary choice for the majority of patients with WM. There is a crucial requirement for prospective phase III trials in WM.
- Mateos M.V. Waldenström's disease. 22nd Congress of the European Hematology Association; 2017 June 22–25; Madrid, Spain.
- Treon S.P. Molecular Biology of Waldenström's disease. 22nd Congress of the European Hematology Association; 2017 June 22–25; Madrid, Spain.
- Kastritis E. Novel treatment strategies in Waldenström's Macroglobulinemia. 22nd Congress of the European Hematology Association; 2017 June 22–25; Madrid, Spain.

Understanding your specialty helps us to deliver the most relevant and engaging content.
Please spare a moment to share yours.
Please select or type your specialty
 Thank you
Thank youRelated articles
Newsletter
Subscribe to get the best content related to lymphoma & CLL delivered to your inbox