All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a healthcare professional. If you are a patient or carer, please visit the Lymphoma Coalition.
The Lymphoma Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the Lymphoma Hub cannot guarantee the accuracy of translated content. The Lymphoma Hub and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


The Lymphoma & CLL Hub is an independent medical education platform, sponsored by AbbVie, BeOne Medicines, Miltenyi Biomedicine, Nurix Therapeutics, Roche, Sobi and Thermo Fisher Scientific and supported through independent educational grants from Bristol Myers Squibb, Incyte, Lilly, and Pfizer. Funders are allowed no direct influence on our content. The levels of sponsorship listed are reflective of the amount of funding given. View funders.
Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account to access:
Bookmark & personalize site content
Receive alerts for new content in your areas of interest
View lymphoma & CLL content recommended for you
Understanding the mutational landscape of plasmablastic lymphoma: a genomic analysis
Plasmablastic lymphoma (PBL) is a rare and aggressive B cell malignancy, often associated with immunosuppression, refractoriness to chemotherapy, and poor outcomes. PBL is characterized by plasmacytic differentiation markers and association with Epstein-Barr virus (EBV). Around 50% of PBL cases harbor MYC translocations, usually rearranging MYC to the heavy chain immunoglobulin locus. However, these genetic aberrations and their role in the molecular pathogenesis of PBL is not well understood. A comprehensive understanding of the mutational landscape in patients will help to improve outcomes in patients with PBL. Frontzek, et al.1 recently published in Nature Communications, a comprehensive genome-wide cohort study investigating the molecular pathogenesis of PBL.
Methods
Overall, 96 formalin-fixed, paraffin embedded tissue samples were collected from cases of primary PBL; diagnosis was confirmed via central review by expert hematopathologists, and all cases fulfilled the World Health Organization 2017 diagnostic classification criteria.
Several methods were employed in this genome wide study, including:
- Immunohistochemistry for selected markers, including IRF4 and Ki-67.
- Fluorescence in situ hybridization to determine the frequency of MYC translocations.
- WES was used for library generation, sequencing and alignment, quality control, basic and advanced variant filtering, and gene level mutation analysis.
- Determination of somatic copy number alterations (SCNAs).
- Western blot, short hairpin ribonucleic acid (shRNA) library screen and cell viability assay.
Results
Molecular pathogenesis
In total, 86% (82/95) of PBL cases did not express the B-cell antigen CD20 while the remaining 14% (13/95) showed weak and inconsistent expression. Latently EBV-infected tumor cells were seen in 57% (55/96) of all cases, while 33% (17/52) of those with available HIV infection status were HIV positive.
EBV positive PBL cases did not demonstrate any association with special morphologic PBL subtypes. Fluorescence in situ hybridization was performed using a break-apart rearrangement probe (BAP) in 57 evaluable cases and a MYC-IgH fusion probe (FP) in 63 cases:
- In total, 47% (28/60) of cases harbored a MYC translocation, determined by positivity for BAP and/or FP.
- Thirty five percent of MYC-BAP positive cases were negative for MYC-FP, meaning that in about a third of cases, MYC translocated to a non-IgH partner.
- MYC translocation status was associated with a significantly higher Ki-67 index (p = 8 × 10-4).
Mutational landscape
The RAS-RAF pathway was affected by recurrent mutations with the NRAS oncogene being the most frequently mutated gene (Figure 1).
- All NRAS variants were missense mutations, with all but one occurring at the known hotspot residues (p.G12, p.G13, and p.Q61).
- NRAS and KRAS mutations were mutually exclusive; all BRAF mutations (6%) were in the kinase protein domain.
- Overall, 47% (40/85) of PBL samples displayed a RAS or BRAF mutation, and the majority of mutations were clonal, indicating their role as initial oncogenic driver genes.
There were recurrent mutations activating the JAK-STAT pathway, with STAT3 mutations occurring in 25% of PBL cases (Figure 1).
- Approximately 50% of detected STAT3 mutations were clonal, highlighting their role as cancer candidate genes.
- Interestingly, STAT3 mutations were only observed in 10% of HIV negative patients vs 47% of patients with HIV (p = 0.003), indicative of a pathogenic role of STAT3 mutations, especially in HIV-associated PBL.
- Gene aberrations were not mutually exclusive, as 29% of PBLs with STAT3 mutations harbored concomitant alterations of JAK1-3, SOCS1/3, or PIAS3.
TP53 mutations were also frequent (Figure 1), clustering particularly in the DNA binding domain (exons 5-8). However, only 27% of TP53 mutations were clonal, suggesting a later pathogenic event.
- TP53 mutations were observed in 6% vs 24% of EBV positive and negative PBL cases, respectively (p = 0.011).
- In 70% of samples with TP53 mutation and available copy number status, loss of the alternate allele leading to biallelic inactivation was detected.
Genes affecting the NOTCH signaling pathways were observed in 26% of the samples and included mutations in SPEN (8%), NOTCH1 (7%), NOTCH4 (6%), DTX1 (3%), NOTCH2 (1%), and NOTCH3 (1%).
Figure 1. Frequency of mutations in PBL samples*
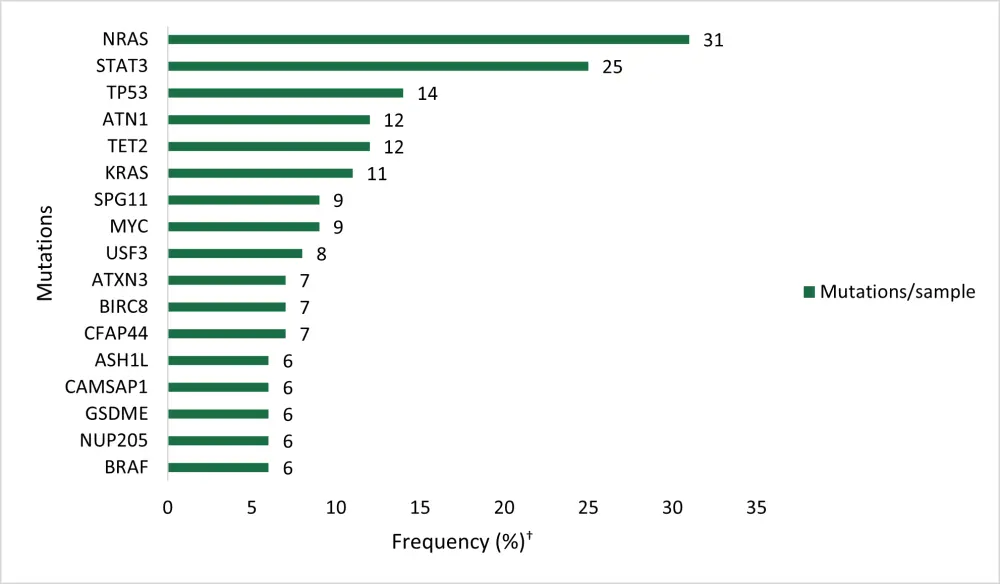
†WES in primary PBL cases (n = 85)
*Adapted from Frontzek, et al.1
Recurrent somatic copy number alterations in PBL
Overall, 82 PBL samples were scanned for copy number alterations using Oncoscan, and SCNAs were identified using GISTIC v2.0.23. Arm-level amplifications were seen in chromosomes 1q, 7p, and 7q and were detected in 42%, 32%, and 33% of PBL cases, respectively. Recurrent arm-level deletions affected chromosomes 13q, 17p, 18p, and 18q and were detectable in 17%, 26%, 18%, and 16% of cases, respectively.
An amplification of 1q23.1 was most specific, identified in 61% of cases, and a wider amplification of 1q23.1 that affected 60 genes was identified in 52% of samples. The antiapoptotic gene MCL1 was identified within this aberration and represents a therapeutic molecular target.
An amplification of 8q24.13 containing TRIB1 was identified in 32% of cases. This is known to induce MED1/ERK signaling and is also amplified in acute myeloid leukemia.
An amplification of 17q22 containing MS12 was present in 21% of cases. MS12 is also overexpressed in acute myeloid leukemia, where is it thought to contribute to poor survival. Deletions at 1p22.1 affecting the gene encoding the potential tumor suppressor RPL5 were identified in 24% of cases and focal deletions of 4q35.2 and 6q26 were identified in 26% and 25% of cases involving the tumor suppressor genes FAT1 and PRKN, respectively.
IPI and EBV status dictate survival in patients with PBL
Corresponding clinical data was available for 49 PBL patients; of those 49 patients, 34 were male, the median age was 62 years, and 82% were treated with a CHOP-like regimen. Nine patients showed a high-risk International Prognostic Index (IPI), 19 an intermediate-risk IPI, and 13 a low-risk IPI. At a median follow up of 21.5 months, 2-year overall survival (OS) was 61%. High-risk IPI patients showed poor 2-year OS of only 11%.
Lymphoma-specific survival (LSS) was significantly inferior in patients who were EBV negative compared with those who were EBV positive (p = 0.002). Significantly inferior outcomes were observed in patients with TP53 mutated PBL compared to patients with TP53 wild type (WT) (p = 0.035). Similarly, NRAS mutations were disposed towards less favorable LSS (p = 0.062) while MYC translocation, MYC expression, and HIV infection had no association with LSS.
Genetic heterogeneity in PBL
Profiles of recurrent mutations were compared with SCNAs for biologically defined subgroups to understand the genetic heterogeneity in PBL (Table 1).
- STAT3 mutations were significantly more frequent in HIV positive compared with HIV negative patients, while recurrent amplification and deletions did not differ significantly.
- Although MYC was the only gene to be significantly mutated, mutations of CFAP44 occurred significantly more frequently in PBL arising in the oral cavity (p = 0.0016).
Table 1. Mutations and SCNAs*
|
Amp, amplification; del, deletion; EBV, Epstein Barr virus; SCNAs, somatic copy number alterations. |
||
|
Mutations and SCNAs |
% |
p value |
|---|---|---|
|
EBV negative vs positive – del |
||
|
1p22.1 |
46 vs 11 |
0.0007 |
|
MYC translocated vs non-translocated - amp |
||
|
1q43 |
58 vs 21 |
0.0122 |
|
2q31.3 |
46 vs 14 |
0.0019 |
|
11q23.3 |
50 vs 18 |
0.0046 |
|
11q25 |
46 vs 14 |
0.0122 |
|
12p11.22 |
42 vs 14 |
0.0060 |
|
Immunocompetent vs immunocompromised patients - del |
||
|
4q35.2 |
50 vs 11 |
0.0035 |
|
18p |
33 vs 4 |
0.0083 |
IRF4 and STAT3 pathways in a plasmablastic cell model
shRNA directed against MYC, IRF4, and STAT3 were among the most significantly depleted. Transduction of the IRF4 shRNAs induced cytotoxicity in PBL-1 cells and in the diffuse large B-cell lymphoma (DLBCL) cell lines OCI-Ly10 from the activated B-cell subtype, but not in germinal center B-cell-like DLBCL cell lines (which were used as negative controls), confirming an addiction to IRF4 signaling in PBL-1 cells.
- Treatment of PBL-1 cells with lenalidomide significantly downregulated IRF4 expression and induced toxicity in PBL-1 cells, suggesting that lenalidomide could be used to treat PBL.
- The STAT3 antisense oligonucleotide AZD9150 induced significant toxicity in PBL-1 cells but not in DLBCL control cells.
- STAT3 WT increased STAT3 signaling to a significantly lesser degree compared with p.D661Y STAT3 mutation.
- In contrast to STAT3 WT, p.Q643R STAT3 mutations partially salvaged PBL-1 cells from STAT3 shRNA-induced toxicity.
- pSTAT3 significantly decreased in the absence of interleukin-6 (IL-6), and in the presence of the pan-JAK inhibitor tofacitinib, decreasing the viability of the cells and suggesting a therapeutic use in PBL.
Conclusion
This comprehensive genetic analysis in a cohort of primary PBL samples of all subtypes has identified previously unknown genetic alterations affecting the RAS-RAF, JAK-STAT, MCL1, IRF4, and NOTCH pathways, particularly JAK-STAT and IRF4, as therapeutically targetable susceptibilities. Collectively, these preclinical findings warrant further research to improve outcomes in patients with PBL.
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content
Your opinion matters
What is the primary reason you use bridging therapy in patients with DLBCL awaiting CAR T-cell therapy?