All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a healthcare professional. If you are a patient or carer, please visit the Lymphoma Coalition.
The lym Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the lym Hub cannot guarantee the accuracy of translated content. The lym and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


The Lymphoma & CLL Hub is an independent medical education platform, sponsored by AbbVie, BeOne Medicines, Miltenyi Biomedicine, Nurix Therapeutics, Roche, Sobi, and Thermo Fisher Scientific and supported through educational grants from Bristol Myers Squibb, Lilly, and Pfizer. Funders are allowed no direct influence on our content. The levels of sponsorship listed are reflective of the amount of funding given. View funders.
Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account and access these new features:
Bookmark content to read later
Select your specific areas of interest
View lymphoma & CLL content recommended for you
EHA 2017 | New approaches in indolent Non-Hodgkin Lymphoma
At the 22nd Congress of the European Hematology Association (EHA), Madrid, Spain, the first session the LH attended was an Educational Session titled “New approaches in indolent Lymphoma”. This session was chaired by Pauline Brice from Hôpital Saint-Louis, Paris, France.
Molecular profiling of indolent Lymphoma (iNHL)
The first talk in the session was presented by Stefano A. Pileri from the European Institute of Oncology, Milan, Italy. He began by explaining that “indolent Lymphoma” is not a term that is used in the WHO classification; it is the clinical definition of a B-cell derived lymphoid neoplasm characterized by a clinical course measurable in years or decades, slow growth, difficult curability, and a tendency to transform into a more aggressive form in some patients. It was emphasized that Gene Expression Profiling (GEP) has rapidly expanded our knowledge of Lymphoma. Following this, various iNHL subtypes were looked at individually in more detail.
Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL)
It appears that slightly more patients harbor unmutated IGHV (50–70%) compared to mutated IGHV (30–50%). Furthermore, approximately one-third of patients have stereotyped B-Cell Receptors (BCRs); this finding had large implications on therapy and prognosis and led to the European Research Initiative on CLL (ERIC) proposing new strategies. Epigenetic analysis for CLL poses a problem as there are two major groups – naïve and memory B-cells, respectively. The main cytogenetic abnormalities in CLL were also summarized in respect to presence or absence of mutated IGHV (Kröbber et al.):
|
*significant difference between cases with and without IGHV mutation |
||
|
|
Frequency |
|
|---|---|---|
|
Aberration |
Mutated IGHV n=132 (44% of cases) |
Unmutated IGHV n=168 (56% of cases) |
|
Clonal aberrations |
80% |
84% |
|
Del(13q)* |
65% |
48% |
|
Isolated del(13q)* |
50% |
26% |
|
Trisomy 12 |
15% |
19% |
|
Del(11q)* |
4% |
27% |
|
Del(17p)* |
3% |
10% |
|
Del(17p) or del(11q)* |
7% |
35% |
It was emphasized that CLL has been reported to carry other mutations such as NOTCH1, SB3FB1, TP53, ATM, BIRC3, POT1, and MYD88. Previously, attempts have been made to combine cytogenetic and mutational data into prognostic groups with little success so far.
Mantle Cell Lymphoma (MCL)
A small group of patients present with a form of MCL that behaves indolently; non-nodal presentation, mainly hypermutated IGHV, lack genomic complexity, lack SOX11 expression, and have excellent outcomes that potentially can be managed with more conservative strategies than conventional disease (Fernàndez et al. Cancer Res. 2010).
The MCL35 assay was discussed, which measures the proliferation signature in RNA derived from routinely available Formalin Fixed Paraffin Embedded (FFPE) biopsies. The assay contains a 17-gene proliferation signature, and yielded expression of adequate quality to assign an assay score and risk group to 108/110 (98%) of archival FFPE biopsies. The assay assigned 26% of patients to the high-risk (median OS, 1.1 years), 29% to the standard-risk (median OS, 2.6 years), and 45% to the low-risk (median OS, 8.6 years; P < 0.001). In multivariate analysis, these risk groups and MIPI were independently associated with OS (P < 0.001 for both). A concordance of risk assessment of 100% was found between three independent laboratories (Scott et al. JCO. 2017). Pros and cons of this assay were discussed in an Educational Session on MCL at the 2017 ASCO Annual Meeting by Jonathon Brett Cohen, MD, MS, read more here.
Lymphoplasmacytic Lymphoma (LPL)/Waldenström Macroglobulinemia (WM)
In 2012, Steven P. Treon, MD, PhD, et al. reported that MYD88 L265P is a frequently harbored in patients with WM and the mutation could aid distinguishing WM and non-IGM LPL from B-cell disorders with other features (NEJM. 2012). However, Pileri cautioned that Next-Generation Sequencing (NGS) studies (Rossi Blood. 2014) have found MYD88 mutations in other mature B-cell malignancies such as CLL and Splenic Marginal Zone Lymphoma (sMZL), as well as in a proportion of Activated B-Cell-Like (ABC) Diffuse Large B-Cell Lymphomas (DLBCLs). Although, identification of MYD88 mutation remains useful as it informs the use of novel drugs.
Hairy Cell Leukemia (HCL)
HCL carries a distinctive gene expression profile. Often, aberrant genes are present resulting in deregulated adhesion (TIMP1, TIMP-4, RECK, CCR7), morphology (LSP1, F-actin, Gas7), phagocytosis (annexin A1, c-Maf), fibrosis (IL-3αR, FLT3), and therapy (IfαR). Overexpression of Annexin A1 has thus far only been observed in HCL and not any other lymphoma. Pileri discussed a diagnostic assay using immunostaining with a specific anti-annexin A1 monoclonal antibody; this method was simple, inexpensive, and highly sensitive and specific (100%) for HCL (Fallini et al. Lancet. 2004).
Pileri moved on to focus on BRAF mutations in HCL. Enrico Tiacci, MD, et al reported (NEJM. 2011) that the BRAF V600E mutation was observed in all 48 HCL patients they evaluated with Whole-Exome Sequencing (WES). More recently in 2012, Tiacci et al. reported a simple, inexpensive, genetics-based test to diagnose HCL in whole-blood samples of patients. The test detects BRAF V600E through a sensitive allele-specific PCR qualitative assay followed by agarose-gel electrophoresis. The test identified BRAF V600E in all 123 HCL samples investigated containing as little as 0.1% leukemic cells. The mutation was detected at multiple time-points during the course of disease, including post-therapy which indicates it may have a crucial role in contributing to pathogenesis and maintenance of the leukemic clone (Blood. 2012). Moreover, an immunohistochemistry based assay using a monoclonal antibody to BRAF V600E (VE1) has been tested and found 2/11 post-treatment bone marrow trephine biopsy samples (all 11 patients had achieved CR by conventional staining) harbored isolated cells or foci of cells still expressing BRAF V600E. Therefore, the VE1 antibody could detect Minimal Residual Disease (MRD) below the levels of detection with conventional histochemical reagents in some patients after therapy (Akarca et al. BJH. 2013).
These findings are extremely relevant as BRAF mutation presents as a therapeutic target; for example, vemurafenib has been found to be active in patients with Relapsed/Refractory (R/R) HCL (Tiacci et al. NEJM. 2015).
Splenic Marginal Zone Lymphoma (sMZL)
sMZL has been reported to lack recurrent chromosomal translocations such as those harbored in other types of Lymphoma. Around one-third of sMZLs show a heterozygous deletion in 7q, rarely observed in other Lymphomas. The genes affected by del(7q) remain unknown. Moreover, gain of 3q has been observed in a large subset of cases. Pileri then went into some detail of the pathways which regulate development of sMZL, such as activation of NOTCH2 (Rossi et al. J Exp Med. 2012).
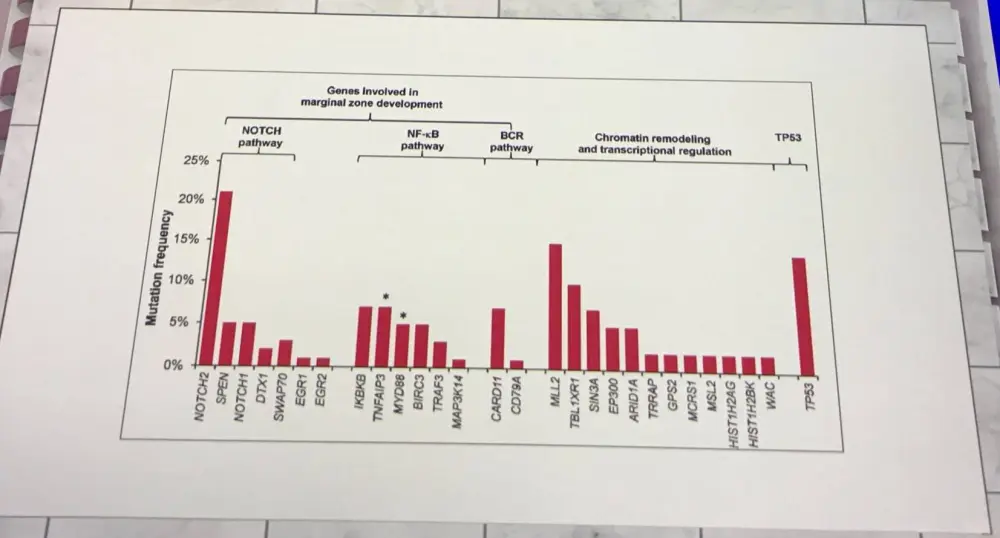
It was stated that the most common somatic change in sMZL is mutation of KLF2 and identifies a subset of patients with a distinct genotype characterized by numerous multi-genetic changes (Clipson et al. Leukemia. 2015). Once again, Pileri warned that this genetic aberration has also been observed in other B-Cell Lymphomas such as HCL, Follicular Lymphoma (FL), and MCL.
Nodal Marginal Zone Lymphoma (nMZL)
nMZL was the next subtype to be discussed and thus far has not been found to harbor del(7q). In nMZL, there is a predominance of mutated IGHV3 and IGHV4 family members, especially IGHV4-34. Similar to sMZL and extranodal MZL of Mucosa-Associated Lymphoid Tissue (MALT), nMZL harbors gains of chromosomes 3 and 8, as well as loss of 6q23–24. Enriched gene expression of gene sets identifying interleukins, integrins, CD40, PI3K, NF-kB, and TGF-β has been reported. nMZL and sMZL share a common mutational profile. One different between them is PTPRD lesions, among the most common alterations in nMZL and found to be absent in sMZL. PTPRD lesions are a novel marker for nMZL and support the distinction between nMZL and sMZL (Spina et al. Blood. 2016).
Extranodal Marginal Zone Lymphoma of Mucosa-Associated Lymphoid Tissue (MALT)
In a screen of 252 primary MALT Lymphomas for translocations t(11;18)(q21;q21), t(14;18)(q32;q21), and t(1;14)(p22;q32), and trisomies 3 and 18 occurred mutually exclusively and were detected in 13.5%, 10.8%, and 1.6% of cases. Trisomy 3 and/or 18 occurred in 42.1%. The frequency at which the translocations occurred varied markedly with the primary site of disease. Trisomies 3 and 18 each occurred predominantly in intestinal and salivary gland MALT Lymphomas (Streubel et al. Leukemia. 2004).
Pileri also discussed abnormalities of TNFAIP3 on chromosome 6q23, which have been observed in 15–30% of cases and can include deletions, mutations, and promoter methylation. TNFAIP3 aberrations are not exlusive to MALT Lymphoma, and have been observed in numerous types of Non-Hodgkin Lymphoma (NHL). Also, MYD88 L265P has been seen in approximately 6–9% of MALT Lymphoma patients.
Follicular Lymphoma (FL)
The translocation t(14;18)(q32;q21) has been observed in around 85–90% of FLs resulting in BCL2/IGH (translocated, non-functional allele). This is not the cause of FL on its own; 4–6 additional mutations are required for full FL, such as: del(1p36), gain of chromosome 3 or 12, gain of p-arm of chromosome 7, gain of q-arm of chromosome 12, breaks at 3q27 (BCL6 rearrangment), or breaks at 6p25 (IRF4 rearrangement).
In terms of FL precursors, it has been reported (Mamessier et al. Haematologica. 2014) that 50–70% of healthy individuals how circulating B-lymphocytes harboring t(14;18). It has been explored whether t(14;18) could be used as a predictive biomarker. Clonal analysis of t(14;18) was carried out in paired pre-diagnostic blood and tumor samples; demonstrated that progression to FL occurred from t(14;18)-positive committed precursors. Also, healthy participants at enrolment who developed FL ≤15 years after demonstrated a markedly higher t(14;18) prevalence and frequency than controls (P < 0.001). A 23-fold higher risk of subsequent FL in blood samples associated with a frequency >10-4 (Odds Ratio [OR], 23.17; 95% CI, 9.98–67.31; P < .001) was found. Risk estimates remained high and significant up to 15 years before diagnosis (Roulland et al. JCO. 2014).
Phylogenetic analysis of serial tumor samples identified CREBBP mutation as an early event and enriched in tumor cell progenitors. These mutations act by permitting immune evasion. CREBBP mutations are an attractive therapeutic target in FL.
Update on Follicular Lymphoma: time beyond chemotherapy?
The second talk in this session was given by Kai Hübel from the University of Cologne, Germany. Chemotherapy-free treatment for indolent Lymphoma was described by Hübel as “the debate of the decade”. So can we eliminate chemotherapy as a treatment for FL? The answer at the moment is “not yet”, but we are nearly there.
CHOP has been the backbone of Lymphoma therapy for the last 40 years. FL survival improved after rituximab (R; immunochemotherapy) became routine therapy; at this point, FL became considered as a more chronic disease. The STiL NHL 1-2003 trial of R-bendamustine challenged R-CHOP and after 117 months of follow-up, time to next treatment was not yet reached with R-B compared to 56.0 months for R-CHOP (77 vs. 109 salvage events, respectively). Therefore, less chemotherapy does not necessarily mean less efficacy (Rummel et al. JCO. 2017). Data of the STiL NHL 1-2003 trial was presented at the 2017 ASCO Annual Meeting (abstract #7501), read more here.
Currently, prognosis of patients with FL is dependent on:
- Clinical features (FLIPI, FLIPI2)
- Genetic mutations (m7-FLIPI)
- Cells of the microenvironment (T-cells [cytotoxic and helper], dendritic cells, and macrophages can influence/repress Lymphoma cells)
- Time of first relapse (less than or more than 2 years)
- Radiological criteria (PET)
However, treatment decision is currently based on histological grading, clinical staging, and tumor burden. Subgroups of patients may need different treatment approaches. The introduction of targeted therapies into current treatment algorithms requires that they are more effective than immunochemotherapy alone, especially in patients with poor prognosis, and that they are less toxic than immunochemotherapy, particularly in patients with favorable outcomes.
Group I: targeting the cell surface
Hübel summarized numerous agents that target molecules on the surface of Lymphoma cells:
|
Target |
Agent |
|---|---|
|
CD20 (type I antibody) |
Rituximab, ofatumumab, ocrelizumab, ublituximab, veltuzumab |
|
CD20 (type II antibody) |
Obinutuxumab |
|
CD22 |
Epratuzumab, inotuzumab ozogamicin |
|
CD79b |
Polatuzumab vedotin |
|
CD19 |
Coltuximab ravtansine |
|
CD37 |
Otlertuzumab |
|
CD80 |
Galiximab |
|
HLA-DR |
IMMU-114 |
|
CD3/CD19 |
Blinatumomab |
The prospective, randomized GAUSS trial comparing the safety and efficacy of obinutuzumab (1,000mg) with rituximab (375mg/m2) in 175 patients with relapsed CD20+ indolent Lymphoma was discussed (Sehn et al. JCO. 2015). Patients who responded also received obinutuzumab or rituximab maintenance therapy every 2 months for up to 2 years. Among patients with FL (n=149), ORR appeared higher for obinutuzumab than rituximab (44.6% vs. 33.3%; P = 0.08). PFS in FL was also higher with obinutuzumab than rituximab (HR, 0.93; 95% CI, 0.60–1.44; P = 0.74). Results of the GALLIUM trial of R-chemo versus O-chemo in frontline FL (updated results presented at ICML 2017, abstract #107, read more here) and the GADOLIN trial of obinutuzumab in rituximab-refractory FL patients (read more here) were highlighted. So what can we learn from these three trials? Well, MRD response potentially identifies new prognostic risk groups and CD20 antibodies are not comparable; obinutuzumab plus chemotherapy was more effective than rituximab plus chemotherapy. It remains uncertain whether chemotherapy is required for optimal effect of these antibodies.
Lastly, in this part of Hübel’s talk, a phase I trial of blinatumomab in 76 patients with R/R Lymphoma (28 FL patients) was discussed. The Maximum Tolerated Dose was 60μg/m2/day and achieved an ORR of 80% in the FL cohort. Neurologic events were dose limiting; grade 3 was reported in 22% of patients (Goebeler et al. JCO. 2016).
Group II: intracellular and epigenetic targets
A list of some of these agents and their targets was presented:
|
Target |
Agent |
|---|---|
|
PI3K |
Idelalisib (δ), duvelisib (δγ), copanlisib (αδ) |
|
BTK |
Ibrutinib, acalabrutinib |
|
BCL-2 |
Venetoclax |
|
Syk |
Entospletinib, fostamatinib |
|
HDAC |
Vorinostat |
|
Proteasome |
Bortezomib, carfilzomib |
|
mTOR |
Temsirolimus, everolimus |
|
MDM2 |
Idasanutlin |
|
EZH2 |
Tazemetostat |
The speaker highlighted results from the phase II DELTA trial of idelalisib monotherapy for R/R iNHL (NCT01282424; Gopal et al. NEJM. 2014). Earlier this year, G. Salles et al. published findings from their FL sub-group analysis, read more here. Idelalisib monotherapy was effective and had an acceptable and manageable safety profile in heavily pretreated patients with R/R FL. In a further subgroup analysis specifically in FL patients who experienced early Progressive Disease (PD) after chemoimmunotherapy (early-early PD ≤12 months n=17; late-early PD 12–24 months n=20), ORR was 56.8% (21/37), with 5 CRs (13.5%) and 16 PRs (43.2%). Response rates were not significantly different between patients with PD ≤12 months after initial therapy (ORR = 70.6%) and patients who experienced PD 12–24 months after initial therapy (ORR = 45.0%; P = 0.18). Median DoR for all 37 patients was 11.8 months (95% CR, 3.8 to not reached). Median PFS was 11.1 months overall, 8 months for PD ≤12 months, and 13.6 months for PD 12–24 months after initial treatment patients. It was concluded that this subgroup analysis suggests that idelalisib may provide an alternative treatment option for patients with double-refractory FL with early relapse after initial therapy (Gopal et al. Blood. 2017). Despite these results, idelalisib carries numerous concerns about its toxicity such as: Pneumocystis jirovecii pneumonia, CMV reactivation, diarrhea, ALT/AST elevation, pneumonitis, and neutropenia. Idelalisib is manageable, but requires careful mandatory monitoring for opportunistic infections and immune toxicities.
The DYNAMO trial (NCT01882803) of duvelisib in patients with double-refractory indolent Non-Hodgkin Lymphoma (iNHL) was covered; updated results were presented at ICML 2017 (abstract #058) by P.L. Zinzani, MD, PhD, read more here.
Ibrutinib was also discussed in some detail. A phase II trial (Fowler et al. ASH. 2016. Abstract #1804) of 60 patients found ibrutinib combined with rituximab achieves an ORR of 85% with a CR of 35% and a PR of 50%. 1-year PFS was 87% and 1-year OS was 98%. Grade 3 or higher AEs included rash (5%), fatigue (7%), and pyrexia (3%). Moreover, results from the phase II DAWN trial (NCT01779791) of ibrutinib in 110 early-relapsed FL was covered (Gopal et al. ASH. 2016. Abstract #1217). An ORR of 20.9% was achieved with a CR rate of 10.9% and SD achieved in 30.9% of patients. Median DoR was 19.4 months, median PFS was 4.6 months, and 2-year OS was 63%. Grade 3 or higher AEs included diarrhea (9%) and fatigue (14%).
Results from the first-in-human phase I trial of venetoclax in R/R NHL was also discussed, which the LH reported on in January 2017; read more here. Venetoclax was shown to be an active single agent treatment option with favorable toxicity profile; combination with other therapies may result in greater efficacy.
Tazemetostat was the last agent in this group to be focused on. In a phase II that included 29 patients with FL, ORR was 63% and 28% in patients with mutated and wild-type EZH2. Grade 3 AEs were reported in 18% of patients, and AEs that occurred in >10% of patients included nausea, thrombocytopenia, cough, diarrhea, fatigue, and asthenia. While at ICML 2017, we interviewed Dr Michael Dickinson from the Peter MacCallum Cancer Center, Victoria, AUS, on the use of tazemetostat in FL and DLBCL; watch it here.
Group III: targeting the microenvironment
Selected drugs in this last group were also summarized:
|
Target |
Agent |
|---|---|
|
Immunomodulation |
Lenalidomide |
|
PD-1 |
Nivolumab, pembrolizumab, pidilizumab |
|
PD-L1 |
Atezolizumab, durvalumab |
|
KIR |
Lirilumab |
|
CD137 |
Urelumab |
Hübel discussed results from a phase II study of lenalidomide combined with rituximab that included 50 patients with untreated FL, of which 46 were evaluable (Fowler et al. Lancet Oncol. 2014). CR was achieved in 40 (87%) patients and 5 (11%) had a PR. The most common grade 3 or 4 AEs among all patients (n=110) in the trial were neutropenia (35%), muscle pain (9%), rash (7%), cough, dyspnoea, other pulmonary symptoms, fatigue, and thrombosis (5% each), as well as thrombocytopenia (4%). Results of the phase III RELEVANCE trial, based on this phase II data, are eagerly awaited.
The phase IIIb MAGNIFY trial (NCT01996865) of lenalidomide plus rituximab in R/R FL was also covered. Results were recently presented at the 2017 ASCO Annual Meeting (abstract #7502) by David Jacob Andorsky, MD, which the LH reported on; read more here. It was concluded that induction with R2 followed by maintenance demonstrated efficacy and was well tolerated in double-refractory and early relapse patients.
The efficacy of immune checkpoint blockade in R/R FL was then summarized:
|
Agent |
n |
ORR |
CR |
|---|---|---|---|
|
10 |
40% |
10% |
|
|
32 |
66% |
52% |
|
|
15 |
80% |
60% |
Conclusion
Hübel hypothesized about the next steps in front-line therapy for FL, and emphasized that we need to distinguish indolent from aggressive disease at time of diagnosis and not relapse.
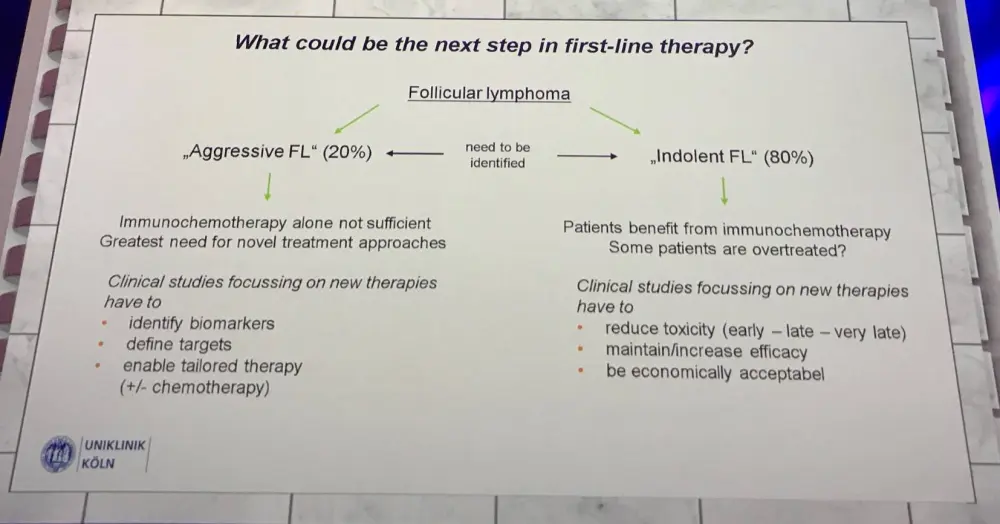
Immunochemotherapy is still the standard of care in front-line and first relapse FL. Efficacy, toxicity, quality of life, and cost of new approaches all have to be compared in long-term follow-up with existing regimens. There is a clear need for the identification of predictive markers enabling individual tailoring of therapy for subgroups of patients.
Treatment of extranodal Marginal Zone B-cell Lymphomas
The last talk in this educational session was presented by M. Raderer from the Medical University of Vienna, AUS.
There is a close association of MALT Lymphoma with infections, such as Helicobacter pylori with gastric MZL. In a Vienna Series 1999–2017, 3/402 MALT Lymphomas were positive for Hepatitis C Virus (0.7%).
Post-treatment evaluation of gastric MALT Lymphoma was summarized (Copie-Bergman et al. Gut. 2003):
|
Score |
Lymphoid infiltrate |
Lymphoepithelial lesions |
Stromal changes |
|---|---|---|---|
|
Complete histological remission (CR) |
Absent or Scattered plasma cells and small lymphoid cells in the lamina propria |
Absent |
Normal or Empty lamina propria and/or fribrosis |
|
Probable minimal residual disease (pMRD) |
Aggregates of lymphoid cells or Lymphod nodules in the lamina propria/muscularis mucosa and/or submucosa |
Absent |
Empty lamina propria and/or fibrosis |
|
Responding residual disease (rRD) |
Dense, diffuse or nodular, extending around glands of the lamina propria |
Focal lymphoepithelial lesions or absent |
Focal empty lamina propria and/or fibrosis |
|
No change (NC) |
Dense diffuse or nodular |
Present ‘may be absent’ |
No changes |
The first-line treatment of all gastric MALT Lymphomas is H. pylori eradication therapy independent of the stage. Despite this, staging must be undertaken before starting eradication therapy and patients who respond should not receive any other therapy (Ruskoné-Fourmestraux et al. Gut. 2011).
It has been reported that bone marrow involvement is not associated with the clinical outcomes of gastric MALT Lymphoma. Overall CR rate was 85.2% during a median follow-up period of 42 months (IQR, 23–66 months) and did not differ between patients with or without bone marrow involvement (78.6% and 85.7%, P = 0.280). First-line eradication therapy was administered to 18/28 (64.3%) of patients with bone marrow involvement, and CR was achieved in 13 patients (72.2%). Logistic regression analysis showed that age and location in the upper part of the stomach were factors related to remission failure (Gong et al. Scand J Gastroenterol. 2016).
Furthermore, it is recommended that H. pylori negative patients with gastric MALT Lymphoma can also undergo anti-H. pylori treatment (Ruskoné-Fourmestraux et al. Gut. 2011). In a study of clarithromycin eradication, out of 12 patients, 4 achieved a CR and 2 achieved a PR (Raderer et al. Ann Haematol. 2014). We discussed the use of clarithromycin in extranodal MZL with Prof. Andrés Ferreri at ICML 2017; watch it here.
When going further than antibiotics, for localized disease radiotherapy is recommended. Both radiotherapy and chemotherapy have a curative potential in localized gastric MAL Lymphoma. There is no recommendation in favor of either of these two modalities. If clinical trials are available, then patients should be put forward. Other recommendations include systemic treatment and radio-immunotherapy for both localized and disseminated disease (Ruskoné-Fourmestraux et al. Gut. 2011).
An overview of therapy with 90Y-ibritumomab tiuxetan (Zevalin) was given (Zizani et al. Best Pract Res Clin Haematol. 2017):
|
Author |
Setting |
n |
ORR |
CR |
PFS |
Toxicity |
|---|---|---|---|---|---|---|
|
Esmaeli |
OAL de novo |
9 |
89% |
78% |
- |
Thrombocytopenia, anemia |
|
Hoffmann |
MALT relapsed |
6 |
83% |
67% |
- |
Neutropenia, thrombocytopenia |
|
Vanazzi |
MALT relapsed |
30 |
90% |
77% |
NR |
Neutropenia, thrombocytopenia |
|
Lossos |
MALT de novo |
11 |
16% |
88% |
46.6 |
Neutropenia, thrombocytopenia |
|
Samniego |
MZL de novo |
11 |
100% |
- |
81.8 |
Neutropenia, thrombocytopenia |
Therapy for MALT Lymphoma with anti-CD20 antibodies is an encouraging area; for example, ofatumumab (O-MA1 trial) achieved a response rate of 81% and a CR of 50%. After follow-up of 25.1 months, only 1 relapse was reported.
Following this, the IELSG-19 phase III trial comparing chlorambucil alone or in combination with rituximab as first-line therapy for MALT Lymphoma was covered. We reported the final results of this trial in March of this year, read more here. Chlorambucil in combination with rituximab was more effective in treating MALT patients than monotherapy with either drug.
A chemotherapy-free approach (lenalidomide plus rituximab) for MALT was also discussed:
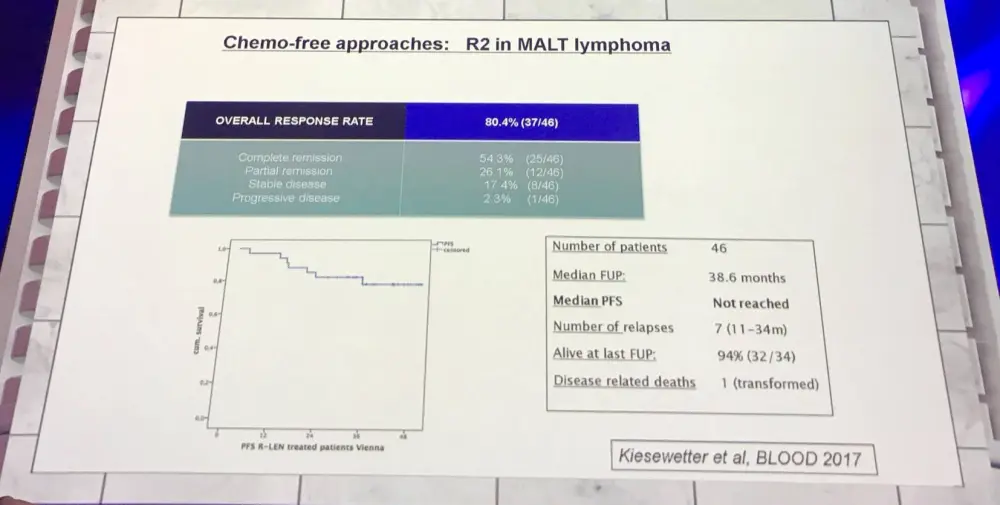
Raderer concluded the talk with four take home messages:
- pylori eradication is standard for gastric MALT Lymphoma
- Ocular adnexal MALT lymphoma may also be treated with antibiotics upfront (doxorubicin achieved an ORR of 38–66%)
- Both radiotherapy and systemic therapy may be applied to localized disease
- The optimal systemic regimen remains to be defined
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content
Your opinion matters
In your experience, what is the average time to secure a reimbursed CAR T-cell therapy manufacturing slot for patients with DLBCL (from decision to treatment with a CAR T-cell therapy)?