All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a healthcare professional. If you are a patient or carer, please visit the Lymphoma Coalition.
The Lymphoma Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the Lymphoma Hub cannot guarantee the accuracy of translated content. The Lymphoma Hub and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


The Lymphoma Hub is an independent medical education platform, sponsored by Roche, Sobi, AbbVie, BeOne, Miltenyi Biomedicine, Thermo Fisher, Nurix Therapeutics and Caribou Biosciences and supported through independent educational grants from Incyte, Bristol Myers Squibb, Lilly and Pfizer. Funders are allowed no direct influence on our content. The levels of sponsorship listed are reflective of the amount of funding given. View funders.
Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account to access:
Bookmark & personalize site content
Receive alerts for new content in your areas of interest
View lymphoma & CLL content recommended for you
Genetic dysregulation of B-cell lymphoma: A focus on epigenetic modifiers in DLBCL and FL from ESH 2020
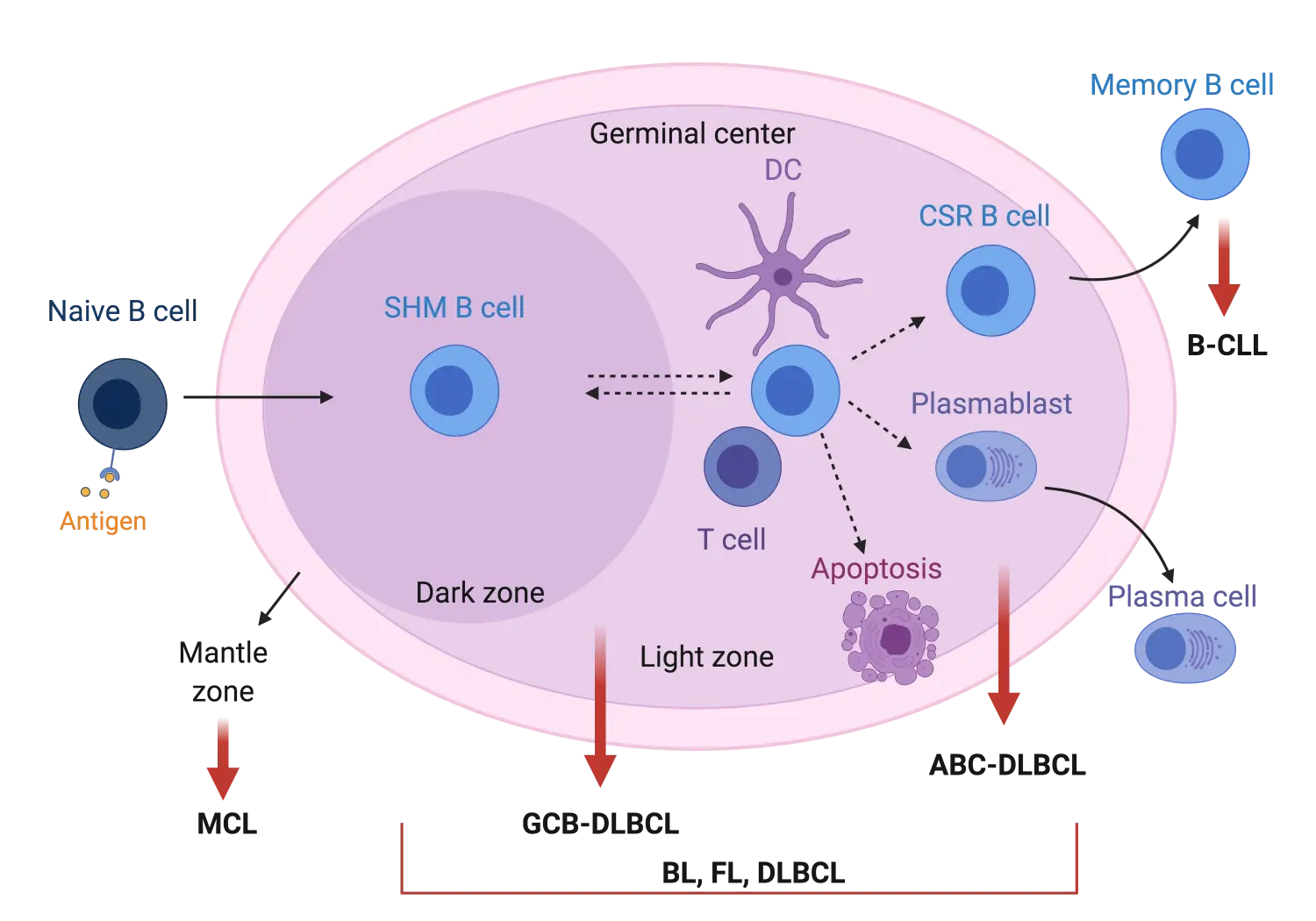
The majority of lymphomas originate from B cells undergoing neoplastic changes (90–95%) and these are classified as B-cell lymphomas.1 Most B-cell cancers arise from the reactions occurring within lymphoid germinal centers (GC). Upon antigen stimulation of naïve B cells, GC are formed in the respective secondary lymphoid tissues and the GC reaction starts (Figure 1). Antigen-stimulated B cells will initially proliferate and undergo somatic hypermutation (SHM) of their immunoglobulin (Ig) variable genes in order to achieve better Ig affinity against the antigen. These processes occur in the dark zone of the GC. B cells will also move to the so-called light zone where interaction with neighbouring dendritic and T cells will determine whether they are selected to become memory B cells, plasma cells or undergo apoptosis. B cells can move bidirectionally between the light and dark GC zones if further rounds of SHM and proliferation are required (Figure 1).1,2,3,4
Most of the non-Hodgkin lymphoma (NHL) subtypes originate from B cells undergoing GC reactions as shown in Figure 1. More specifically, follicular lymphoma (FL) and the GC B-cell-like subtype (GCB) of diffuse large B-cell lymphoma (DLBCL) molecularly resemble B cells of the light zone, while the activated B-cell-like (ABC) DLBCL subtype resembles plasma cell precursors (plasmablasts). Burkitt lymphoma (BL) has been shown to possess molecular characteristics of B cells in either the dark or light GC zone. Tumor cells in B-cell chronic lymphocytic leukemia (B-CLL) show high similarity to memory B cells, while in mantle cell lymphoma (MCL), tumor cells arise from B cells within the mantle zone of the GC (Figure 1).3,4
Figure 1. Schematic representation of germinal center B-cell reaction upon antigen stimulation2
With the great advances in genomic sequencing, numerous genetic modifications have been identified for each lymphoma subtype. We hereby provide the most commonly associated genetic alterations in DLBCL by cell-of-origin (COO) subtype (GCB, ABC; Table 1), in FL, MCL, BL, and classical Hodgkin lymphoma (cHL; Table 2). The effect and prognostic impact of these genetic modifications have been widely studied for each lymphoma subtype. In this article, we will focus on genetic alterations of epigenetic modifiers that have been identified specifically in DLBCL and FL. This topic was further discussed by two leading investigators in the field, Laura Pasqualucci and Riccardo Dalla-Favera, during the 2020 European School of Haematology (ESH) e-conference.3,4 We hereby summarize the latest updates on genetic-driven epigenetic dysregulation in DLBCL and FL, and the potential for drug targeting strategies.
Table 1. Most common genetic modifications observed among the COO DLBCL subgroups2
|
ABC, activated B-cell-like; amp, amplification; COO, cell-of-origin; GCB, germinal center B-cell-like; d, deletion; DLBCL, diffuse large B-cell lymphoma; m, mutation; Tx, chromosomal translocation. |
||
|
GCB- and ABC-DLBCL |
GCB-DLBCL |
ABC-DLBCL |
|---|---|---|
|
BCL6 Tx |
BCL2 Tx/m |
TNFAIP3 m/d |
|
*MLL2/MLL3 m |
GNA13 m |
MYD88 m |
|
*CREBBP/EP300 m/d |
*EZH2 m |
CDKN2A/B d |
|
B2M/CD58 m/d |
TNFRSF14 m |
BCL2 amp |
|
TP53 m |
BCL6/BSE1 m |
PRDM1 m/d |
|
MEF2B m |
MYC Tx |
CD79A/B m |
|
FOXO1 m |
PTEN d |
CARD11 m |
Table 2. Most common genetic modifications in HL, FL, MCL and BL1
|
amp, amplification; BL, Burkitt lymphoma; d, deletion; FL, follicular lymphoma; HL, Hodgkin lymphoma; m, mutation; MCL, mantle cell lymphoma; tFL, transformed FL; Tx, chromosomal translocation. |
|||
|
FL/tFL |
HL |
MCL |
BL |
|---|---|---|---|
|
BCL2 Tx |
JAK1 m |
CCND1 Tx |
MYC Tx |
|
*MLL2 m |
STAT3 m |
ATM m/d |
ID3 m |
|
*CREBPP m |
STAT5B m |
TP53 m |
TCF3 (E2A) m |
|
*EZH2 m |
JAK2 m |
*WHSC1 m |
CCND3 m |
|
*HIST1H1E m |
PTPN1 m |
NOTCH1/2 m |
— |
|
MEF2B m |
SOCS1 m/d |
BIRC3 m/d |
— |
|
STAT6 m |
†LMP2A |
MEF2B m |
— |
|
SOCS1 m |
— |
— |
— |
|
PAPLOG amp |
— |
— |
— |
|
REL amp |
— |
— |
— |
|
MYC amp |
— |
— |
— |
|
TP53 d |
— |
— |
— |
|
TNFAIP3 d |
— |
— |
— |
Novel GC B-cell subpopulations can inform DLBCL outcomes
The traditional compartmentalization of GC B cells into dark or light zone B cells based on their different transcriptional profiles has enabled the identification of two COO DLBCL subtypes (GCB, ABC). These subtypes have been shown to predict DLBCL outcomes following standard chemotherapy, with GCB conferring a better survival than ABC.4 Nevertheless, recent single cell analysis has revealed that GC B cells are far more heterogeneous than originally believed. In fact, 13 transcriptionally distinct GC B-cell subpopulations were identified by Holmes et al. (2020)5 that can be classified into dark zone, intermediate zone, light zone, plasmablasts or memory precursors in the order of progression within the GC reaction. The authors showed that these new COO signatures are also capable of predicting outcomes in DLBCL patients. Although in need of further validation, their results showed that when combining the GCB/ABC classification with the new single-cell signatures, poor prognosis subgroups exist within the ‘favorable’ GCB subtype, and conversely, good prognosis subgroups exist within the ‘less favorable’ ABC subtype.4,5 This new subcategorization might lead to even better outcome prediction and more personalised treatment management for DLBCL patients.
Gene alterations in GC B-cell epigenetic modifiers (DLBCL and FL)
For these heterogenous GC B cells to be able to exist and to rapidly switch between these highly diverse transcriptional profiles without leading to malignancies, they require high and well-orchestrated epigenetic plasticity. In fact, the evidence presented below shows that desynchronisation of epigenetic regulation in these cells contributes to the development of B-cell lymphomas, like DLBCL and FL.3 This information provides a new therapeutic avenue for B-cell lymphoma.
Approximately all FL cases and a significant proportion of DLBCL patients harbor genetic alterations in epigenetic regulators like EP300, CREBPP, EZH2, MLL2 (KMT2D) or HIST1H1E (Tables 1 and 2). These cause aberrant epigenetic regulation in GC B cells and contribute at least partly to disease development. In support of that, MLL2 or CREBPP deletion in animal models leads to the development of FL and DLBCL only when combined with BCL2 dysregulation, as observed in patients. BCL2 dysregulation alone had a much lower tumorigenic phenotype in the same models.3,4
Studies have shown that CREBPP or MLL2 mutations occur early during the evolution of lymphoma in a common precursor cell (CPC) before its clonal expansion. In the case of FL, these early mutations occur before the clonal evolution towards FL or transformed FL (upon acquisition of additional genetic alterations).3,4 This is a significant finding as further investigation of these early epigenetic alterations in the tumor CPC may identify novel therapeutic targets aimed at inhibiting early lymphomagenesis and elimination of the original clone.
CREBPP is a histone acetyltransferase, while MLL2 is a histone methyltransferase. Both enzymes have been shown to occupy large genomic regions that seem to have GC enhancer/super-enhancer profiles. Epigenetic modulation by MLL2 in these regions has been shown to regulate genes required for the light zone GC reaction, like interferon response, immune signalling, B-cell receptor signalling, and G-protein signalling. Similarly, CREBPP also modulates the expression of genes involved in the light zone GC reaction, by inhibiting the actions of BCL6, the main suppressor of light zone gene expression (dark zone modulator).
CREBPP and MLL2 mutations are inactivating events leading to aberrant epigenetic modulation and improper GC cell signature acquisition.3 Moreover, the detected CREBPP and MLL2 mutations in DLBCL/FL are mainly monoallelic, indicating that the cells require at least some basal level of acetyltransferase/methyltransferase activity in order to survive.3 This finding has now been further explored as a potential target against tumor cells. CREBPP is structurally and functionally very similar to EP300 and they are known to compensate for each other when one is lost or malfunctional. Moreover, although CREBPP and EP300 mutations have been identified separately in DLBCL and FL cases, they never occur together. Based on these findings, the team of Laura Pasqualucci generated mice with conditional double knockout of both CREBPP and EP300 specifically in GC B cells.3 Animals with combined loss of these two epigenetic modulators were completely incapable of forming GC upon antigen stimulation, unlike their heterozygous counterparts. This CREBPP/EP300 dependency was further confirmed in CREBPP-mutant DLBCL cells upon inhibition of EP300 with small molecule inhibitors (CU329, CCS1477).3 Such agents hold great therapeutic potential and encourage the development of EP300-specific inhibitors for further clinical evaluation in DLBCL and FL.
Conclusions
The discovery of genetic alterations in B cell malignancies, like DLBCL and FL, has played a crucial role in the better understanding of the disease and its pathogenesis. Such knowledge enables the exploitation of genetic targets for therapeutic or prognostic purposes. More specifically, epigenetic mutations seem to be the hallmark of GC-derived B-cell malignancies, with CREBPP/MLL2 mutations having been identified as early events in the lymphomagenesis cascade. The data summarized above provide strong evidence in favor of new treatments that target such epigenetic modulators with the aim of eliminating the original tumor clone and inhibiting lymphomagenesis early. One druggable target that fulfils such criteria seems to be EP300.
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content
Your opinion matters
In patients with R/R LBCL who progress after CAR‑T, which of the following data would most strengthen your confidence in considering BV+R2?