All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a healthcare professional. If you are a patient or carer, please visit the Lymphoma Coalition.
The Lymphoma Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the Lymphoma Hub cannot guarantee the accuracy of translated content. The Lymphoma Hub and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


The Lymphoma & CLL Hub is an independent medical education platform, sponsored by AbbVie, BeOne Medicines, Caribou Biosciences, Miltenyi Biomedicine, Nurix Therapeutics, Roche, Sobi and Thermo Fisher Scientific and supported through independent educational grants from Bristol Myers Squibb, Incyte, Lilly, and Pfizer. Funders are allowed no direct influence on our content. The levels of sponsorship listed are reflective of the amount of funding given. View funders.
Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account to access:
Bookmark & personalize site content
Receive alerts for new content in your areas of interest
View lymphoma & CLL content recommended for you
The role of the tumor microenvironment on CAR T-cell therapy in B-cell lymphoma
Question 1 / 1
Which of the following factors within the tumor microenvironment (TME) are not associated with limiting the antitumor response of CAR T-cell therapy?
A
Good nutrient supply
B
Low oxygen levels
C
Immunosuppressive cells
D
Dense extracellular matrix and fibrotic stroma
While chimeric antigen receptor (CAR) T-cell therapies are clinically effective, there are persisting challenges including response variability, resistance, and side effects.
The tumor microenvironment (TME) is a complex internal environment where tumors are located; they are comprised of various components such as tumor cells, stromal cells, immune cells, extracellular matrix (ECM), and soluble factors that can influence CAR T-cell function and behavior.1
The Lymphoma Hub previously reported on the tumor intrinsic mechanisms of resistance to CAR T-cell therapy in patients with lymphomas. Here, we summarize a recently published review article by Cai et al. in European Journal of Hematology,1 on the mechanisms by which the TME promotes efficacy or resistance to CAR T-cell therapy in B-cell lymphoma, as well the emerging strategies modulating the TME to improve CAR T-cell efficacy and improve patient outcomes.
The role of the TME in B-cell lymphoma
The pathogenesis of B-cell lymphoma involves interactions between tumor cells and the TME. The TME has two main components: the immune microenvironment, consisting of immune cells (regulatory T cells [Tregs], tumor-associated macrophages [TAMs], myeloid-derived suppressor cells [MDSCs], tumor-associated neutrophils, natural killer cells, dendritic cells); and the non-immune microenvironment, consisting of fibroblasts, ECM, pericytes, mesenchymal stromal cells, and stromal cells. The mechanism by which the immune components exert immunosuppressive effects has been summarized in Figure 1.
Figure 1. Role of the immune cells in the TME*
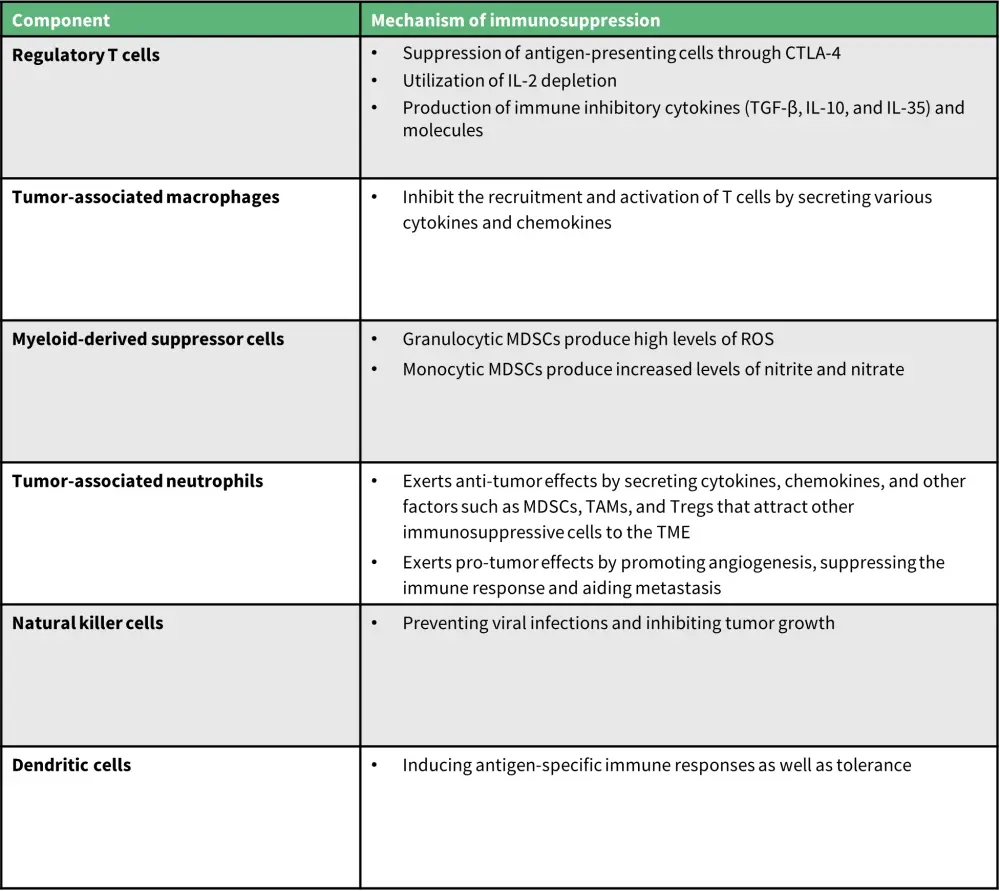
IL, interleukin; MDSC, myeloid-derived suppressor cell; ROS, reactive oxygen series; TAM, tumor-associated macrophage; TGF-β, transforming growth factor beta; TME, tumor microenvironment; Treg, regulatory T cell.
*Data from Cai, et al.1
The role of the TME in CAR T-cell therapy
Key mechanisms through which the TME can either promote efficacy or resistance to CAR T-cells are depicted in Figure 2.
Figure 2. Mechanisms involved in the impact of TME on CAR T-cell response*
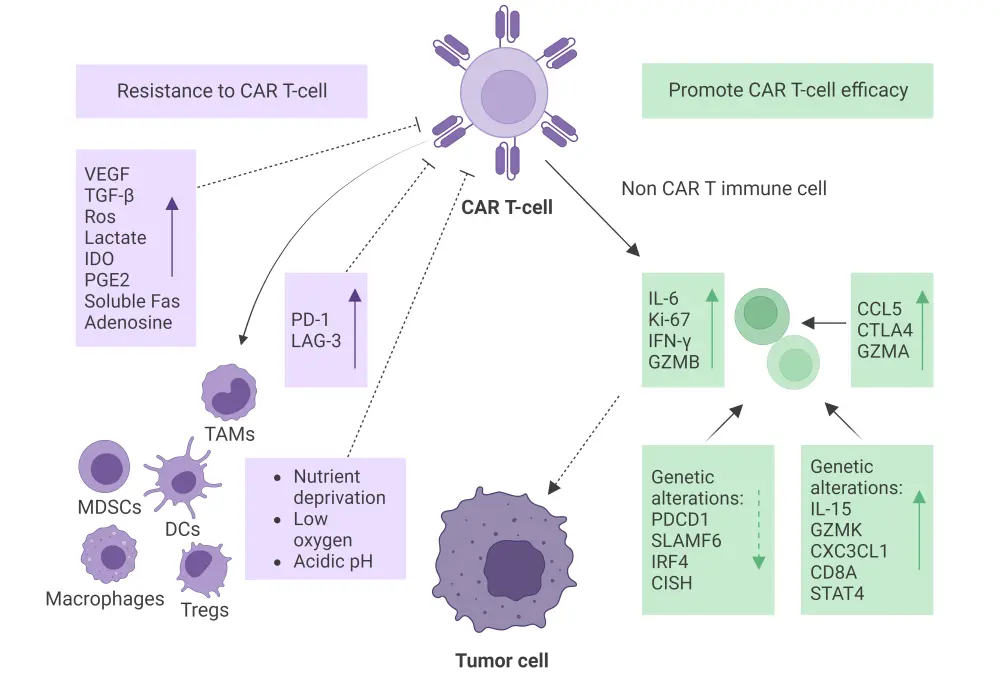
CAR, chimeric antigen receptor; CCL5, chemokine C-C motif ligand 5; CD8A, cluster of differentiation 8A; CISH, chromogenic in situ hybridization; CTLA4, cytotoxic T-lymphocyte associated protein 4; CXC3CL1, chemokine C-X3-C motif ligand 1; DC, dendritic cell; IDO, indoleamine 2,3-dioxygenase; IFN-γ, interferon gamma; IL, interleukin; IRF4, interferon regulatory factor 4; Ki-67, kiel-67; LAG-3, lymphocyte activation gene 3; MDSC, myeloid-derived suppressor cell; PD-1 and PDCD1, programmed cell death protein 1; PGE2, prostaglandin E2; SLAMF6, signaling lymphocytic activation molecule family member 6; STAT4, signal transducer and activator of transcription 4; TAM, tumor-associated macrophage; TGF-β, transforming growth factor beta; TME, tumor microenvironment; Treg, regulatory T cell; VEGF, vascular endothelial growth factor.
*Data from Cai, et al.1 Created with BioRender.com.
The role of the TME in promoting CAR T-cell efficacy
Multiplex immunostaining and in situ hybridization assays performed in the ZUMA-1 trial of patients with diffuse large B-cell lymphoma (DLBCL) receiving axicabtagene ciloleucel suggested that CAR T-cells are activated in the TME but only make up a small percentage of the total T cell population ≥5 days post-infusion; and non-CAR immune cells activated in the TME expressed interleukin (IL)-6, kiel-67, interferon-γ and granzyme B at the highest levels in biopsies with CAR T-cells.
A ZUMA-2 trial analysis revealed that the presence of cytotoxic T cells expressing immune checkpoints or co-expressing thymocyte selection–associated high-mobility group box was negatively associated with circulating CAR T-cell levels in the post-treatment TME. Conversely, enrichment of chemokines (chemokine C-C motif ligand [CCL]5 and CCL22), γ-chain cytokines (IL-15, IL-7, IL-21), and interferon-regulated molecules (cluster of differentiation [CD]274, CD276, and cytotoxic T-lymphocyte associated protein-4) in the TME were linked to T-cell activation and activity.
Genetic alterations and mutational profiles of tumor cells within the TME can impact the CAR T-cell response. One analysis of post-CAR T-cell and non-CAR T-cell treatment showed that patients with a prolonged progression-free survival had reduced expression of immune checkpoint genes and genes associated with T cell exhaustion compared with those with a shorter progression-free survival. Likewise, a single-cell sequencing analysis of peripheral blood mononuclear cells in patients with DLBCL receiving CAR T-cell therapy revealed a marked increase in the expression of programmed cell death protein 1 (PDCD1) and signaling lymphocytic activation molecule family member 6 (SLAMF6) on CD8+ cells; it also led to downregulation of interferon regulatory factor 4 (IRF4) and cytokine-induced src homology 2 (SH2) protein involved in preventing CAR T-cell exhaustion and negatively regulating cytokine signal transduction, respectively.
Additionally, a multicenter trial comparing pre- and post-axicabtagene ciloleucel treatment biopsies from 14 patients with relapsed/refractory (R/R) B-cell non-Hodgkin lymphoma (NHL) showed upregulation of the CCL5, cytotoxic T-lymphocyte associated protein-4 (CTLA-4), and granzyme A genes post-treatment involved in immune cell recruitment, immune regulation, and cytotoxic activity, respectively. This study also reported upregulation of IL-15, granzyme K, chemokine (C-X3-C motif) ligand 1 (CXC3CL1), CD8A, and signal transducer and activator of transcription 4 (STAT4) related to T cell proliferation, homing, and effector function, suggesting enhanced T cell activity within the TME.
The role of the TME in resistance to CAR T-cells
Physical barriers, immunosuppressive cells, inhibitory molecules, and immune checkpoints within the TME can contribute to resistance to CAR T-cell therapy. Nutrient deprivation, low oxygen levels, and acidic pH of the TME can negatively impact CAR T-cell survival, proliferation, and effector functions, limiting CAR T-cell activity and contributing to resistance. Additionally, the dense ECM and fibrotic stroma within the TME are physical barriers preventing effective penetration and infiltration of CAR T-cells into the tumor, thus limiting their ability to eliminate the tumor.
A whole-genome sequencing analysis of 51 tumor samples from 49 patients with LBCL treated with CAR T-cell therapy found that the presence of complex structural variants, apolipoprotein B mRNA-editing enzyme, catalytic polypeptide (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide [APOBEC]) mutational signatures, and genomic damage from reactive oxygen species are predictors of CAR T-cell resistance.
Immunosuppressive cells within the TME such as Tregs, MDSCs, and TAMs can inhibit CAR T-cell function, limiting their antitumor response. One CAR T-cell study in patients with DLBCL found that tumor expression of interferon signaling and high levels of M-MDSC were separately associated with a lack of durable response to CAR T-cell treatment. Moreover, the presence of macrophages and dendritic cells in the TME may secrete soluble factors such as vascular endothelial growth factor and transforming growth factor (TGF)-β, which contribute to CAR T-cell resistance through several mechanisms such as the promotion of abnormal tumor vasculature, or facilitation of the anti-inflammatory polarization of immune cells.
Further to this, a phase I clinical study investigating anti-CD19 JWCAR029 showed that patients with R/R B-NHL achieving complete remission before CAR T-cell therapy had lower levels of chemokines that negatively regulate the recruitment of TAMs (CCL2 and chemokine C-X-C motif ligand 8), Tregs, and MDSCs (chemokine C-X-C motif ligand 12, CCL3, CCL4, and CCL5) to TME compared with patients achieving a partial remission. On the other hand, patients achieving a partial remission had increased levels of immunosuppressive cytokines (IL-10 and TGF-β1), MDSCs (CD33 and CD14) and tumor-associated fibroblasts (fibroblast activation protein [FAP], tenascin-C [TNC], chondroitin sulfate proteoglycan 4 [CSPG4], platelet-derived growth factor receptor alpha [PDGFRA], S100 calcium-binding protein A4 [S100A4], asporin [ASPN], stanniocalcin-1 [STC1], and integrin alpha M [ITGAM]) when compared with those achieving complete remission, suggesting that the immunosuppressive nature of the TME before CAR T-cell treatment could influence CAR T-cell response.
Programmed cell death ligand 1 molecules which engage with inhibitory receptors on CAR T-cells are upregulated by tumor cells, resulting in T cell exhaustion and reduced antitumor activity. A multicenter trial in patients with aggressive B-NHL found upregulation of the immune checkpoint molecules programmed cell death ligand 1 and lymphocyte activation gene 3 post-treatment, suggesting a possible immune resistance mechanism.
The role of TME in CAR T-cell toxicity
The TME can also mediate side effects associated with CAR T-cell therapy, including cytokine release syndrome (CRS), neurotoxicity, and on-target off-tumor effects (Figure 3).
Figure 3. Side effects of CAR T-cell therapy mediated by TME*
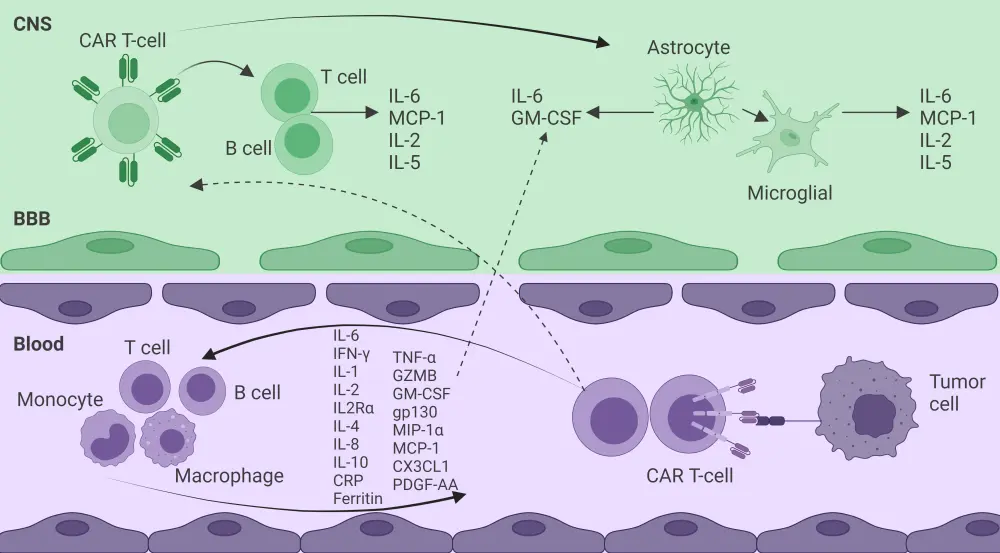
BBB, blood–brain barrier; CAR, chimeric antigen receptor; CNS, central nervous system; CRP, C-reactive protein; CX3CL1, chemokine C-X3-C motif ligand 1; GM-CSF, granulocyte-macrophage colony-stimulating factor; gp130, glycoprotein 130; GZMB, granzyme B; IFN, interferon; IL, interleukin; MCP-1, monocyte chemoattractant protein-1; MIP-1α, macrophage inflammatory protein-1 alpha; PDGF-AA, platelet-derived growth factor-AA;TME, tumor microenvironment; TNF-α, tumor necrosis factor alpha.
*Data from Cai, et al.1 Created with BioRender.com.
CRS is characterized by the release of multiple cytokines and is not only produced by the CAR T-cells but by other immune cells within the TME such as macrophages, T cells, B cells, monocytes, and endothelial cells. Cytokines such as C-reactive protein, ferritin, interferon-γ, IL-2, soluble IL-2 receptor α, IL-6, IL-10, tumor necrosis factor-α, granulocyte/macrophage colony-stimulating factor, and macrophage inflammatory protein-1α contribute to severe CRS and its clinical manifestations of fever, hypotension, and organ dysfunction. A multicenter trial found that levels of CX3CL1, granzyme B, IL-4, IL-6, and platelet-derived growth factor-AA could predict the risk of severe CRS in patients receiving CAR T-cell therapy.
Immune effector cell-associated neurotoxicity syndrome is clinically characterized by neurological symptoms such as encephalopathy, tremors, dysgraphia, apraxia, and seizures caused by interactions between CAR T-cell and target cells that result in the activation and release of inflammatory factors (IL-6, monocyte chemoattractant protein-1, IL-2, and IL-5) in the central nervous system. Moreover, the destruction of the blood–brain barrier may lead to the migration of CAR T-cells to the brain parenchyma causing an increase in cytokine and protein levels that leads to central nervous system inflammation and toxicity.
Strategies modulating the TME to increase CAR T-cell efficacy
Attenuating the TME can improve the efficacy of CAR T-cell therapy and improve the clinical outcomes for patients with B-cell lymphoma. One such strategy to overcome the immunosuppressive effects of TME is by depleting immunosuppressive cells such as MDSCs, TAMS, and Tregs to promote infiltration of immune cells, reduce the impact of the ECM and tumor vascular system, and regulate hypoxia, nutrient deficiency, and metabolic status in the TME.
In a study by Lexus et al.,2 engineered CAR T-cells delivering RN7SL1, an endogenous RNA that activates retinoic acid-inducible gene I/melanoma differentiation-associated gene 5 signaling, resulted in reduced accumulation of MDSCs and TGF-β in myeloid cells. CAR T-cells expressing IL-7 and CCL19 can improve the immune cell infiltration to solid tumors as well as prolong CAR T-cell survival in tumors.
Other approaches that have shown promising results include combining oncolytic viruses with CAR T-cells to induce immunogenic cell death in cancer cells and significantly inhibit tumor growth or combining CAR T-cells with immune checkpoint molecules (programmed cell death protein 1, T-cell immunoglobulin and mucin domain 3, and lymphocyte activation gene 3) to overcome T cell dysfunction caused by inhibitory signals. Programmed cell death protein 1 inhibition with pembrolizumab has demonstrated promising efficacy and safety outcomes in patients with R/R B-NHL following CAR T-cell treatment.
Conclusion
This article highlights the complexity of the TME and how it can influence CAR T-cell function and behavior in B-cell lymphoma as well as emerging strategies aimed at modulating the TME to improve the efficacy of CAR T-cell therapy. Approaches being investigated to optimize CAR T-cell function and improve patient outcomes include combining CAR T-cell therapy with immune checkpoint inhibitors, modifying the TME through targeted therapies, and genetically engineering CAR T-cells to resist inhibitory signals or secrete immunomodulatory molecules.
Future studies evaluating the molecular and cellular events occurring post-CAR T-cell infusion such as TME composition changes, immune cell interactions, cytokine signaling, and potential resistance mechanisms are needed to further develop more effective CAR T-cell therapies and strategies to reduce treatment-related toxicities.
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content
Your opinion matters
In patients with R/R LBCL who progress after CAR‑T, which of the following data would most strengthen your confidence in considering BV+R2?